Approach to hemolytic anemia medicine
Reading File
Finding Sources
Searching PubMed
"hemolytic anemia" diagnosis management
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
I now have all the material needed for a comprehensive response. Here is the complete clinical approach to hemolytic anemia:
Approach to Hemolytic Anemia
Definition and Pathophysiology
Hemolytic anemia is defined as accelerated destruction of red blood cells (RBCs), shortening their normal 120-day lifespan. The resulting tissue hypoxia triggers erythropoietin release, driving marrow erythroid hyperplasia and peripheral reticulocytosis - the hallmarks of any hemolytic process. In severe cases, extramedullary hematopoiesis develops in the liver, spleen, and lymph nodes. - Robbins & Kumar Basic Pathology, p. 384
Step 1 - Confirm Hemolysis (Lab Diagnosis)
The following tests confirm that anemia is hemolytic in origin:
| Test | Finding in Hemolysis | Mechanism |
|---|---|---|
| Reticulocyte count | Elevated (reticulocytosis) | Compensatory marrow response |
| Serum haptoglobin | Low / undetectable | Binds free Hb; complex cleared by macrophages |
| Serum LDH (LDH-1) | Elevated | Released from lysed RBCs |
| Indirect (unconjugated) bilirubin | Elevated (~2-2.5 mg/dL) | Hb breakdown product |
| Peripheral blood smear | Fragmented cells, spherocytes, etc. | Morphologic clue to etiology |
| Serum potassium | May be elevated | Intracellular K released on lysis |
| Carboxyhemoglobin (CO) | Elevated | Released during porphyrin ring oxidation |
Low haptoglobin is the most sensitive individual marker; it falls even in purely extravascular hemolysis because macrophages "regurgitate" enough free Hb to consume haptoglobin. - Henry's Clinical Diagnosis and Management by Laboratory Methods, p. 130
Step 2 - Localize: Intravascular vs. Extravascular
This is a critical early branch point:
| Feature | Extravascular | Intravascular |
|---|---|---|
| Site of destruction | Spleen (macrophages) | Within circulation |
| Mechanism | Reduced deformability, opsonization | Complement fixation, mechanical trauma |
| Hemoglobinemia | Absent | Present |
| Hemoglobinuria | Absent | Present (red/brown urine) |
| Hemosiderinuria | Absent | Present (chronic) |
| Iron deficiency | Not seen | May develop (iron lost in urine) |
| Jaundice | Common | May occur |
| Splenomegaly | Common | Less prominent |
| Examples | Hereditary spherocytosis, warm AIHA | TTP, cold AIHA, G6PD crisis, ABO incompatibility |
Both types share low haptoglobin. - Robbins & Kumar Basic Pathology, p. 384
Step 3 - Classify by Etiology: Inherited vs. Acquired
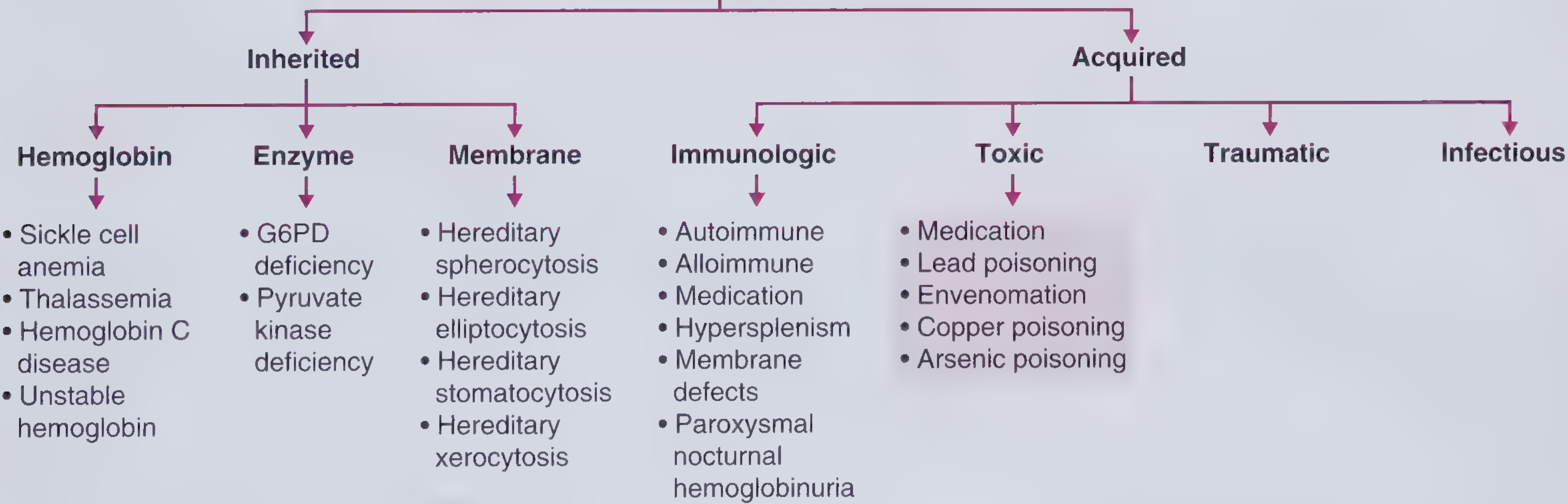
Classification of hemolytic anemia. - Frameworks for Internal Medicine
Clinical clue: Inherited causes tend to present younger, may have a family history (though not always - recessive inheritance or de novo mutations can obscure this). Acquired causes can occur at any age; HUS, for example, is common in children. - Frameworks for Internal Medicine, p. 344
INHERITED CAUSES
A. Hemoglobin Defects (Hemoglobinopathies)
Sickle cell anemia
- Autosomal recessive missense mutation in beta-globin
- Deoxygenated HbS polymerizes, distorting/damaging RBCs
- Moderate-to-severe hemolytic anemia + vaso-occlusive pain crises, stroke, infection risk
- Most prevalent in sub-Saharan Africa and malarial regions
Thalassemia
- Autosomal codominant; alpha- or beta-globin synthesis reduced
- Results in microcytic, hypochromic anemia
- In beta-thalassemia major: unpaired alpha-chains precipitate, causing ineffective erythropoiesis
- Common in Mediterranean and Southeast Asian populations
Others: Hemoglobin C disease, unstable hemoglobin variants
B. Enzyme Defects
G6PD deficiency (most common RBC enzyme defect, affects up to 1/5 of the population in endemic regions)
- X-linked recessive; protective against malaria
- G6PD produces NADPH, which stabilizes antioxidants within RBCs
- Without G6PD, RBCs are susceptible to oxidative stress
- Most patients asymptomatic at baseline; acute hemolysis triggered by:
- Drugs: dapsone, sulfamethoxazole, primaquine, nitrofurantoin
- Foods: fava beans
- Infections
- Smear: Heinz bodies (denatured Hb), "bite cells" (from splenic pitting)
- Heterozygous females can be equally affected due to X-inactivation
Pyruvate kinase (PK) deficiency
- Autosomal recessive; impairs ATP generation in RBCs
- Causes chronic extravascular hemolysis
C. Membrane/Cytoskeleton Defects
Hereditary spherocytosis (most common)
- Autosomal dominant; mutations destabilize the membrane skeleton (spectrin, ankyrin, band 3)
- Loss of membrane lipid bilayer → sphere-shaped, non-deformable RBCs
- Trapped and destroyed in spleen
- Triad: anemia, splenomegaly, cholelithiasis (pigment stones)
- Smear: spherocytes (small RBCs lacking central pallor); positive osmotic fragility test
Hereditary elliptocytosis, stomatocytosis, xerocytosis - less common membrane disorders
ACQUIRED CAUSES
A. Immunologic
Warm AIHA (most common acquired hemolytic anemia in non-malarial countries)
- IgG antibodies react with RBC antigens at body temperature
- Extravascular hemolysis (spleen)
- Causes: primary (idiopathic), SLE, CLL, methyldopa
- Smear: spherocytes
- Diagnosis: Direct Antiglobulin Test (DAT/Coombs) positive
Cold AIHA (Cold Agglutinin Disease)
- IgM antibodies fix complement at low temperatures
- Intravascular hemolysis
- Causes: Mycoplasma pneumoniae (>50% of cases), EBV, CLL
- Symptoms worsen in cold; acrocyanosis
Alloimmune hemolysis
- Alloantibodies after transfusion or pregnancy
- Most dramatic: ABO-incompatible transfusion - IgM activates complement, causing rapid massive intravascular hemolysis → DIC, renal failure, shock, death
Drug-induced immune hemolysis
- Methyldopa: triggers AIHA
- Quinine: triggers TMA (thrombotic microangiopathy)
- Hapten mechanism: penicillin coats RBCs, targeted by drug-dependent antibodies
Paroxysmal Nocturnal Hemoglobinuria (PNH)
- Acquired clonal defect; RBCs lack GPI-anchored complement regulatory proteins (CD55, CD59)
- Complement-mediated intravascular hemolysis
- Classic triad: hemolytic anemia, thrombosis (especially venous), cytopenias
B. Microangiopathic Hemolytic Anemia (MAHA) - Traumatic/Mechanical
RBCs are sheared by fibrin strands or abnormal surfaces:
| Condition | Key Feature |
|---|---|
| TTP | ADAMTS13 deficiency → ultra-large vWF multimers → platelet thrombi in microvasculature. Pentad: MAHA + thrombocytopenia + neurological symptoms + fever + renal failure |
| HUS | Mostly children; Shiga toxin (E. coli O157:H7) or S. pneumoniae; triad: MAHA + thrombocytopenia + AKI. Do NOT use antibiotics (may precipitate HUS) |
| DIC | Intravascular fibrin deposition; MAHA + thrombocytopenia + elevated PT/aPTT + low fibrinogen |
| Prosthetic heart valves | Mechanical shear of RBCs across abnormal valve surfaces |
| Malignant hypertension, eclampsia | Endothelial damage, fibrin deposition |
Smear in all MAHA: schistocytes (helmet cells, RBC fragments) - pathognomonic finding.
Henry's describes a leukoerythroblastic picture when marrow microvasculature is involved (metastatic tumor, myelofibrosis) - releasing nucleated RBCs, myelocytes, and metamyelocytes. - Henry's Clinical Diagnosis and Management by Laboratory Methods
C. Infectious
| Pathogen | Mechanism | Notes |
|---|---|---|
| Malaria | Splenic destruction of infected RBCs (mainly extravascular); Falciparum can cause "blackwater fever" (severe intravascular) | Most common cause of hemolytic anemia worldwide |
| E. coli O157:H7 | Shiga toxin → HUS | Antibiotics contraindicated |
| Mycoplasma pneumoniae | Cold agglutinins (IgM) | Peak weeks 1-4 after infection |
| Clostridium perfringens | Toxin-mediated direct membrane destruction | Mortality ~75%; treat with penicillin + clindamycin |
| Babesia | Direct RBC invasion | Maltese cross tetrad on smear; tick-borne |
| Bartonella bacilliformis | Direct RBC invasion | Endemic to Andes region |
D. Toxic/Chemical
| Agent | Mechanism |
|---|---|
| Dapsone, nitrates, chlorates, cisplatin, methylene blue | Oxidative hemolysis even without G6PD deficiency |
| Lead poisoning | Acquired P5'N-1 deficiency; basophilic stippling on smear |
| Wilson's disease | Copper release from necrotic liver; ~10% of cases |
| Brown recluse spider (Loxosceles reclusa) | Venom causes mixed intravascular/extravascular hemolysis |
| Arsenic poisoning | Direct RBC membrane damage |
E. Hypersplenism
- Spleen traps and destroys otherwise normal RBCs due to massive enlargement
- Pancytopenia pattern common
Step 4 - Key Peripheral Blood Smear Findings
| Morphology | Associated Condition |
|---|---|
| Spherocytes | Hereditary spherocytosis, warm AIHA |
| Schistocytes / helmet cells | MAHA (TTP, HUS, DIC, valve hemolysis) |
| Sickle cells | Sickle cell disease |
| Target cells | Thalassemia, HbC disease |
| Bite cells / Heinz bodies | G6PD deficiency |
| Elliptocytes | Hereditary elliptocytosis |
| Basophilic stippling | Lead poisoning, thalassemia |
| Ring forms (Maltese cross) | Malaria (Falciparum), Babesia |
| Nucleated RBCs + immature WBCs | Myelophthisic / leukoerythroblastic |
Step 5 - Diagnostic Algorithm Summary
Anemia confirmed
↓
Reticulocytosis? + Low haptoglobin? + Elevated LDH?
↓ YES → Hemolysis confirmed
Peripheral blood smear
↓
┌─────────────────────────────────────┐
│ Schistocytes? │
│ → MAHA workup: TTP, HUS, DIC, valve│
│ Spherocytes? │
│ → DAT/Coombs: AIHA vs. HS │
│ Sickle cells? │
│ → Hb electrophoresis │
│ Ring forms? │
│ → Malaria smear, Babesia PCR │
│ Bite cells? │
│ → G6PD assay │
└─────────────────────────────────────┘
↓
Intravascular or Extravascular?
↓
Hemoglobinuria → Intravascular → PNH (flow cytometry), TTP, ABO incompatibility
No hemoglobinuria → Extravascular → Spleen-based destruction
Age/History:
Young + family history → Inherited (HS, G6PD, SCC, Thal)
Older + new onset → Acquired (AIHA, MAHA, drugs, infection)
Management Principles by Cause
| Cause | Treatment |
|---|---|
| Warm AIHA | Corticosteroids (first-line); rituximab, splenectomy (refractory) |
| Cold AIHA | Cold avoidance; rituximab; treat underlying cause |
| TTP | Emergency plasma exchange (PEX); rituximab for acquired TTP |
| HUS (STEC) | Supportive; dialysis if needed; NO antibiotics |
| G6PD crisis | Remove trigger; supportive; transfuse if severe |
| Hereditary spherocytosis | Splenectomy (curative); folate supplementation |
| Sickle cell | Hydroxyurea, pain management, exchange transfusion for crises |
| Drug-induced | Stop the offending drug |
| PNH | Eculizumab (anti-C5 complement inhibitor) |
| Malaria | Antimalarials (artemisinin-based) |
Recent Evidence
Recent reviews from Hematology ASH Education Program (2025) on AIHA management and cold agglutinin disease confirm that rituximab is increasingly used as second-line therapy, and novel complement inhibitors are expanding the treatment landscape for cold AIHA and PNH. The first consensus recommendations for Evans syndrome (AIHA + ITP) were published in Lancet Haematology in 2024.
Sources: Robbins & Kumar Basic Pathology | Frameworks for Internal Medicine | Henry's Clinical Diagnosis and Management by Laboratory Methods | PubMed (PMID 41347987, 41348049, 38968944)
This is a shared conversation. Sign in to Orris to start your own chat.