Myocardial infraction
Reading File
Finding Sources
Searching PubMed
"myocardial infarction"[MeSH Terms] AND management
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
I now have comprehensive information from multiple authoritative sources. Let me compile the full answer.
Myocardial Infarction (MI)
Definition
Myocardial infarction (MI) - commonly called a "heart attack" - is the death of cardiac muscle due to prolonged ischemia, resulting from an imbalance between myocardial oxygen supply and demand. Roughly 800,000 individuals in the United States experience an MI each year (nearly one MI every 40 seconds), causing almost 400,000 deaths annually.
- Robbins, Cotran & Kumar Pathologic Basis of Disease
Classification
| Type | Description |
|---|---|
| STEMI | ST-segment Elevation MI - full-thickness (transmural) ischemia, complete occlusion |
| NSTEMI | Non-ST-segment Elevation MI - partial occlusion, subendocardial involvement |
| Type 1 | Spontaneous MI due to plaque rupture/erosion with thrombosis |
| Type 2 | MI due to supply-demand mismatch (e.g., vasospasm, severe anemia, tachyarrhythmia) |
Epidemiology & Risk Factors
- 10% occur in people < 40 years, 45% occur in people < 65 years
- Through middle age, male sex confers higher relative risk
- Females are generally protected during reproductive years; post-menopausal decline in estrogen leads to worsening CAD - IHD is the most common cause of death in older females
- Post-menopausal hormonal replacement therapy has not been shown to be protective and may be detrimental (pro-thrombotic effect)
Major risk factors: hypertension, dyslipidemia, diabetes mellitus, smoking, obesity, family history, sedentary lifestyle.
Pathogenesis
The sequence of events underlying most MIs:
- Plaque disruption - an atheromatous plaque is eroded or suddenly ruptured by endothelial injury, intraplaque hemorrhage, or mechanical forces, exposing subendothelial collagen and necrotic plaque contents
- Platelet activation - platelets adhere, aggregate, and release thromboxane A₂, ADP, and serotonin, causing further aggregation and vasospasm
- Coagulation cascade - activation by tissue factor adds to the growing thrombus
- Complete occlusion - within minutes, the thrombus can fully occlude the coronary artery lumen
When angiography is performed within 4 hours of MI onset, thrombotic occlusion is demonstrated in ~90% of cases. The left anterior descending artery (LAD) is occluded most often, followed by the right coronary artery (RCA) and left circumflex.
Non-atherothrombotic causes (~10% of MIs)
-
Coronary vasospasm (e.g., cocaine, ephedrine use)
-
Embolism (from mural thrombus, infective endocarditis vegetations, patent foramen ovale)
-
Vasculitis, sickle cell disease, amyloid deposition
-
Hemodynamic compromise (shock, severe hypotension)
-
Robbins, Cotran & Kumar Pathologic Basis of Disease
Pathophysiology
Biochemical cascade of ischemia:
- Seconds - cessation of aerobic metabolism → inadequate ATP and creatine phosphate production → accumulation of lactic acid
- Within 1 minute - loss of myocardial contractility (before cell death)
- 20-40 minutes - irreversible cell injury begins (point of no return)
- 1-2 hours - macroscopic irreversibility without reperfusion
The area at risk = the anatomic region supplied by the occluded artery. The wavefront phenomenon describes how ischemic necrosis begins in the most vulnerable subendocardium (most remote from coronary supply) and progresses outward toward the epicardium over time.
Reperfusion injury can paradoxically worsen cell death through oxidative stress and calcium overload when blood flow is suddenly restored.
Morphological Changes Over Time
(From Robbins - Table 12.5)
| Time | Gross Appearance | Light Microscopy |
|---|---|---|
| 0-30 min | None | None (reversible injury only) |
| 0.5-4 hr | None | Variable fiber waviness at border |
| 4-12 hr | Dark mottling | Early coagulative necrosis, edema |
| 12-24 hr | Dark mottling | Coagulative necrosis, pyknosis, myocyte hypereosinophilia, contraction band necrosis, early neutrophilic infiltrate |
| 1-3 days | Yellow-tan center | Coagulative necrosis, brisk neutrophil infiltration |
| 3-7 days | Hyperemic border, soft center | Neutrophil death, macrophage phagocytosis, early granulation tissue |
| 7-10 days | Maximally yellow-tan and soft | Well-developed granulation tissue, new vessels |
| 2-8 weeks | Gray-white scar | Collagen deposition, decreased cellularity |
| > 2 months | Complete scar | Dense collagenous scar |
- Robbins, Cotran & Kumar Pathologic Basis of Disease
Clinical Features
Symptoms
- Chest pain - the hallmark; typically crushing, pressure-like, or squeezing substernal pain, often radiating to the left arm, left jaw, back, or epigastrium; lasting > 20-30 minutes and not relieved by nitrates
- Dyspnea, diaphoresis, nausea, vomiting
- Silent MI - particularly common in diabetics and elderly (diabetic autonomic neuropathy masks pain); may present as dyspnea, fatigue, or sudden hemodynamic collapse
Signs
- Tachycardia, hypotension (or hypertension early)
- S3/S4 gallop, new mitral regurgitation murmur (papillary muscle dysfunction)
- Signs of heart failure: pulmonary rales, elevated JVP, peripheral edema
- Pericardial friction rub (transmural MI with pericarditis, day 2-3)
ECG Changes
Three major electrical abnormalities occur in acute MI:
| Defect | Mechanism | ECG Change |
|---|---|---|
| Rapid repolarization of infarcted cells | K⁺ channel opening | ST segment elevation |
| Decreased resting membrane potential | Loss of intracellular K⁺ | TQ depression (recorded as ST elevation) |
| Delayed depolarization | Slowed conduction | ST elevation |
Evolutionary ECG changes:
- Hyperacute T waves (minutes) - peaked, tall T waves
- ST elevation (hours) - hallmark of STEMI; leads overlying the infarct show elevation, opposite leads show reciprocal depression
- T wave inversion (hours to days)
- Pathological Q waves (days to weeks) - reflect electrically silent dead tissue; indicate transmural necrosis
Localizing the infarct by ECG:
-
Anterior (LAD): V1-V4
-
Inferior (RCA): II, III, aVF
-
Lateral (LCx): I, aVL, V5-V6
-
Posterior: tall R in V1-V2 with ST depression (reciprocal)
-
Right ventricle (RCA): ST elevation in right-sided leads (V3R, V4R)
-
Ganong's Review of Medical Physiology
Biomarkers
| Marker | Rises | Peaks | Returns to Normal |
|---|---|---|---|
| Troponin I / T | 3-6 hr | 24-48 hr (cTnT up to 14 days) | 7-14 days |
| CK-MB | 3-6 hr | 12-24 hr | 48-72 hr |
| Myoglobin | 1-3 hr | 6-9 hr | 24 hr (early but non-specific) |
Troponin is the preferred biomarker - most sensitive and specific. High-sensitivity troponin (hsTn) allows "rule-in" and "rule-out" in as little as 1-2 hours. Re-elevation of troponin after normalization suggests reinfarction.
Management
Acute Phase - First Response (MONA + Reperfusion)
- Morphine - for pain relief (though evidence for benefit is debated)
- Oxygen - only if SpO₂ < 90% (routine O₂ may be harmful in normoxic patients)
- Nitrates (sublingual/IV nitroglycerin) - for pain and preload reduction; avoid in RV infarction (risk of severe hypotension), and with recent PDE-5 inhibitor use
- Aspirin - 325 mg loading dose immediately; then 75-100 mg daily
Reperfusion Therapy - The Cornerstone of STEMI Management
Goal: Time is myocardium
| Strategy | Indication | Time Target |
|---|---|---|
| Primary PCI (percutaneous coronary intervention) | STEMI, preferred if available | Door-to-balloon < 90 min |
| Fibrinolysis (tPA, streptokinase, tenecteplase) | STEMI if PCI unavailable within 120 min of first medical contact | Door-to-needle < 30 min |
| CABG | Failed PCI, multivessel disease, left main stenosis | Urgent/semi-urgent |
Primary PCI is the preferred reperfusion strategy when performed in a timely fashion by an experienced team.
- Fuster and Hurst's The Heart, 15th Edition
Antithrombotic Therapy
Antiplatelet agents:
- Aspirin - immediate loading (300-325 mg), then lifelong
- P2Y12 inhibitors (Dual Antiplatelet Therapy - DAPT):
- Ticagrelor (180 mg loading, 90 mg BD) - preferred
- Prasugrel (60 mg loading, 10 mg daily) - preferred over clopidogrel post-PCI
- Clopidogrel (600 mg loading, 75 mg daily) - if others contraindicated
- DAPT is continued for 12 months after ACS; duration may be shortened based on bleeding risk
Anticoagulants (acute phase):
- Unfractionated heparin (UFH), enoxaparin, bivalirudin, or fondaparinux
Other Pharmacological Therapy
| Drug | Indication | Notes |
|---|---|---|
| Beta-blockers | All MI without contraindications | Reduce heart rate, limit infarct size, prevent arrhythmias; start within 24 hr |
| ACE inhibitors / ARBs | EF < 40%, anterior MI, hypertension, diabetes | Reduce remodeling, mortality; start early if stable |
| Aldosterone antagonists (eplerenone, spironolactone) | EF < 40% + HF or diabetes | Additional mortality benefit |
| High-intensity statins (atorvastatin 80 mg) | All MI patients | Reduce recurrent events; start immediately |
Recent evidence update: A 2025 meta-analysis (PMID: 39298680) found that beta-blockers for secondary prevention after MI in patients without reduced ejection fraction or heart failure may have limited benefit, challenging the traditional universal use of beta-blockers post-MI in this subgroup.
Complications
Early (hours to days)
-
Arrhythmias - most common complication; ~90% of patients develop some form
- Ventricular fibrillation - greatest risk in the first hour (most dangerous)
- Complete heart block (especially inferior MI with RCA occlusion)
- Sinus bradycardia (inferior MI)
- Atrial fibrillation
-
Cardiogenic shock - LV pump failure; carries high mortality (~50%)
- Defined as hypotension + signs of tissue hypoperfusion despite adequate filling
-
Acute heart failure - pulmonary edema from LV systolic failure
Days to Weeks
-
Myocardial rupture (1-3% of MIs, usually day 3-7)
- Free wall rupture → hemopericardium → cardiac tamponade (usually fatal)
- Ventricular septal rupture → VSD with acute left-to-right shunt
- Papillary muscle rupture → severe acute mitral regurgitation
- Risk factors: first MI, female sex, age > 60, anterior/lateral wall, no prior LVH
-
Pericarditis - fibrinous; appears 2-3 days post-MI (friction rub, pleuritic chest pain)
-
Dressler syndrome (weeks to months) - immune-mediated pericarditis due to anti-myocardial antibodies
-
LV aneurysm - thinning and outward bulging of infarcted wall; predisposes to:
- Mural thrombus → systemic embolization
- Persistent ST elevation
- Refractory heart failure
- Ventricular arrhythmias
-
Mural thrombus - forms on akinetic/dyskinetic endocardium → stroke risk
- Robbins, Cotran & Kumar Pathologic Basis of Disease, Fig. 12.17
Killip Classification (Clinical Severity)
| Class | Features | Mortality |
|---|---|---|
| I | No heart failure | ~5% |
| II | Mild HF (S3, rales < 50% lung fields) | ~10% |
| III | Pulmonary edema | ~20% |
| IV | Cardiogenic shock | ~80% |
Secondary Prevention (Post-Discharge)
- DAPT: Continue for 12 months; aspirin lifelong thereafter
- High-intensity statin: Atorvastatin 80 mg or rosuvastatin 40 mg; target LDL < 70 mg/dL (or < 55 mg/dL in very high-risk)
- ACE inhibitor/ARB: Especially if EF < 40%, hypertension, or diabetes
- Beta-blocker: If EF < 40% or ongoing ischemia
- Cardiac rehabilitation: Strongly recommended
- Lifestyle modification: Smoking cessation, diet, exercise, weight management
Key Images from Textbooks
Here are the gross pathology complications of MI:
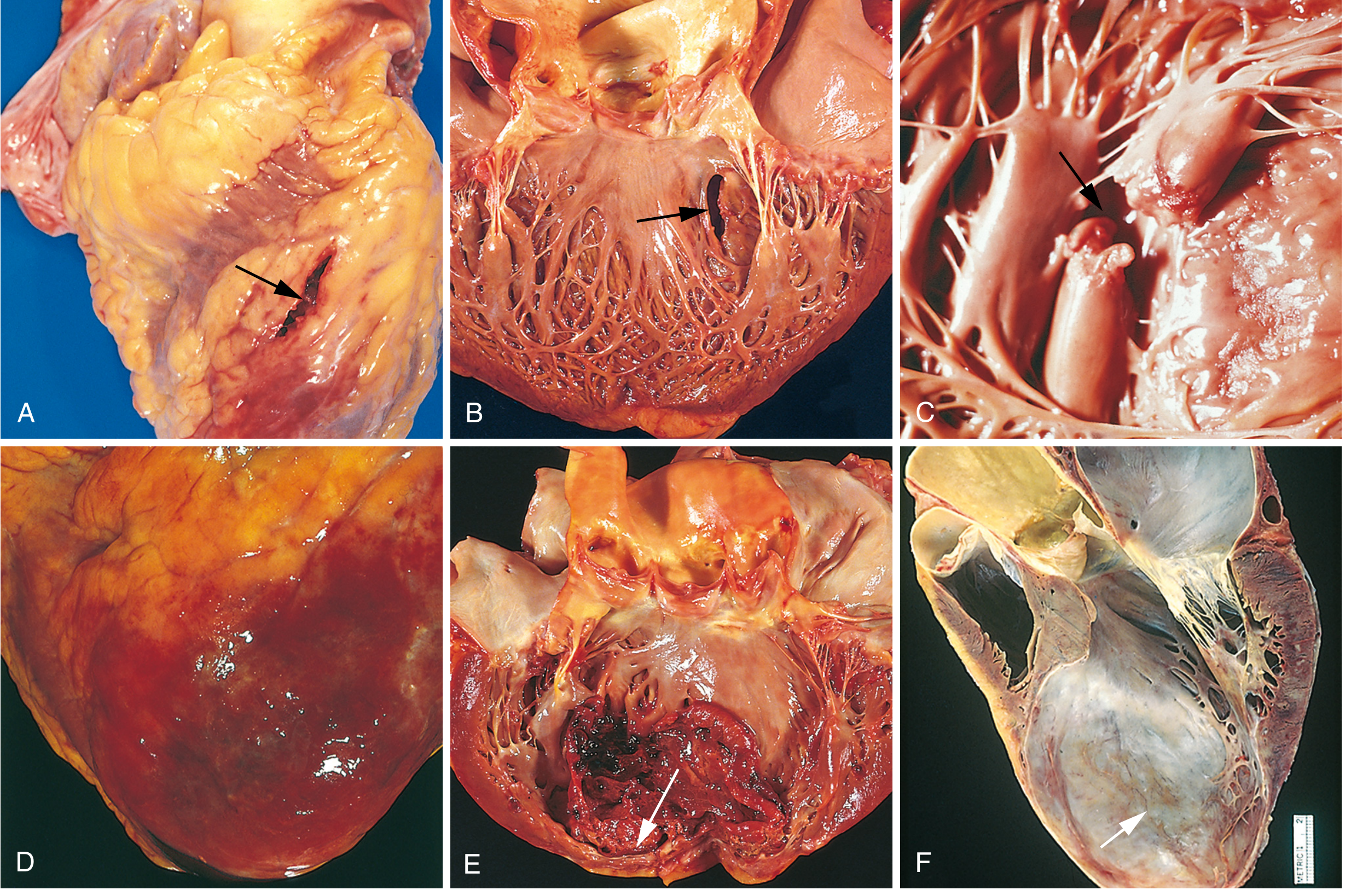
Complications of MI: (A) anterior free wall rupture, (B) ventricular septal rupture, (C) papillary muscle rupture, (D) fibrinous pericarditis, (E) early infarct expansion with mural thrombus, (F) LV aneurysm - Robbins Pathology
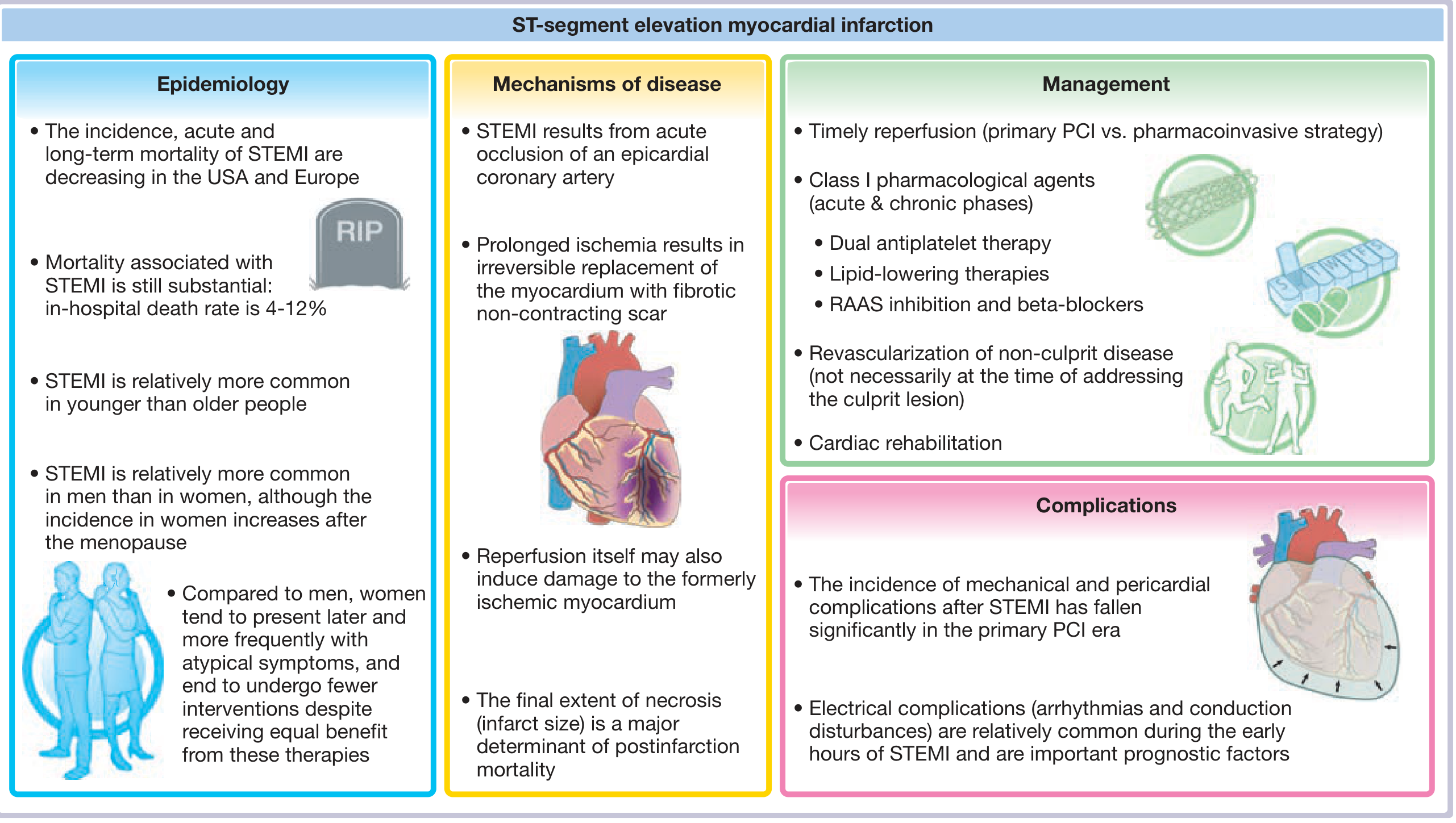
STEMI epidemiology, mechanisms, management and complications overview - Fuster and Hurst's The Heart, 15th Edition
Sources:
- Robbins, Cotran & Kumar Pathologic Basis of Disease (Robbins Pathology), Chapter 12
- Fuster and Hurst's The Heart, 15th Edition, Chapter 18
- Ganong's Review of Medical Physiology, 26th Edition, Chapter 29
- Braunwald's Heart Disease, 2-Vol Set
This is a shared conversation. Sign in to Orris to start your own chat.