Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathophysiology
Searching the Web
ARDS acute respiratory distress syndrome mechanism pathophysiology 2024
Reading File
Reading File
Searching Images
ARDS acute respiratory distress syndrome pathophysiology mechanism diagram
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Reading File
I now have comprehensive material from Harrison's 22E, Murray & Nadel, and Fishman's. I have the key diagram. Let me now compose a thorough answer.
Acute Respiratory Distress Syndrome: Mechanism
ARDS is a syndrome of non-cardiogenic pulmonary edema resulting from diffuse alveolar damage (DAD). Its mechanism spans an initiating insult, a dysregulated inflammatory cascade, structural barrier breakdown, and three overlapping clinical/histological phases.
1. Initiating Insults
Injury to the lung can come from direct or indirect routes:
| Direct (pulmonary) | Indirect (extrapulmonary) |
|---|---|
| Pneumonia (bacterial, viral, fungal) | Sepsis (most common overall cause) |
| Aspiration of gastric contents | Severe pancreatitis |
| Pulmonary contusion | Major trauma / burns |
| Inhalation injury | Blood product transfusion (TRALI) |
| Drowning | Shock / hypoperfusion |
2. The Exudative Phase (Days 0–7): Alveolar-Capillary Barrier Disruption
This is the central pathophysiological event — breakdown of the normally tight alveolar-capillary membrane.
a) Epithelial and Endothelial Injury
The alveolar-capillary unit consists of:
- Type I pneumocytes (95% of alveolar surface): gas exchange; highly vulnerable to injury
- Type II pneumocytes: surfactant synthesis; serve as progenitor cells for repair
- Capillary endothelium: maintains vascular integrity
Direct insults or systemic inflammatory signals activate Toll-like receptors (TLRs) on alveolar type I epithelial cells and resident alveolar macrophages. This triggers secretion of chemokines (IL-8, CXCL1) that recruit circulating immune cells.
b) Neutrophil-Mediated Injury — The Central Effector
Activated neutrophils (PMNs) are the primary mediators of early injury:
- Neutrophil elastase — disrupts intercellular junctions; kills endothelial and epithelial cells; levels in BAL fluid correlate with injury severity
- Matrix metalloproteinases (MMPs) — released by PMNs and macrophages; degrade junctional proteins in epithelia and endothelia
- Reactive oxygen species (ROS) and reactive nitrogen species — overwhelm endogenous antioxidant defenses; cause lipid peroxidation, protein oxidation, and tight junction disruption
- Neutrophil extracellular traps (NETs) — webs of chromatin and antimicrobial proteins that exacerbate epithelial and endothelial injury
- Oxidized phospholipids — damage membrane integrity and surfactant function
Importantly, ARDS can occur even in neutropenic patients, demonstrating that neutrophils are not the sole mediators. — Murray & Nadel's Textbook of Respiratory Medicine
c) Macrophage and Monocyte Contributions
Following initial neutrophilic infiltration, monocytes are recruited and differentiate into macrophages. These secrete:
- TNF-α, IL-1β, IL-6, IL-8 — propagate inflammation
- TRAIL (TNF-related apoptosis-inducing ligand) — induces epithelial apoptosis
- Interferon-β — further immune activation
- Vascular endothelial growth factor (VEGF) — paradoxically increases vascular permeability at high concentrations
Monocyte-platelet aggregates also form, and platelets interact with PMNs to release additional mediators.
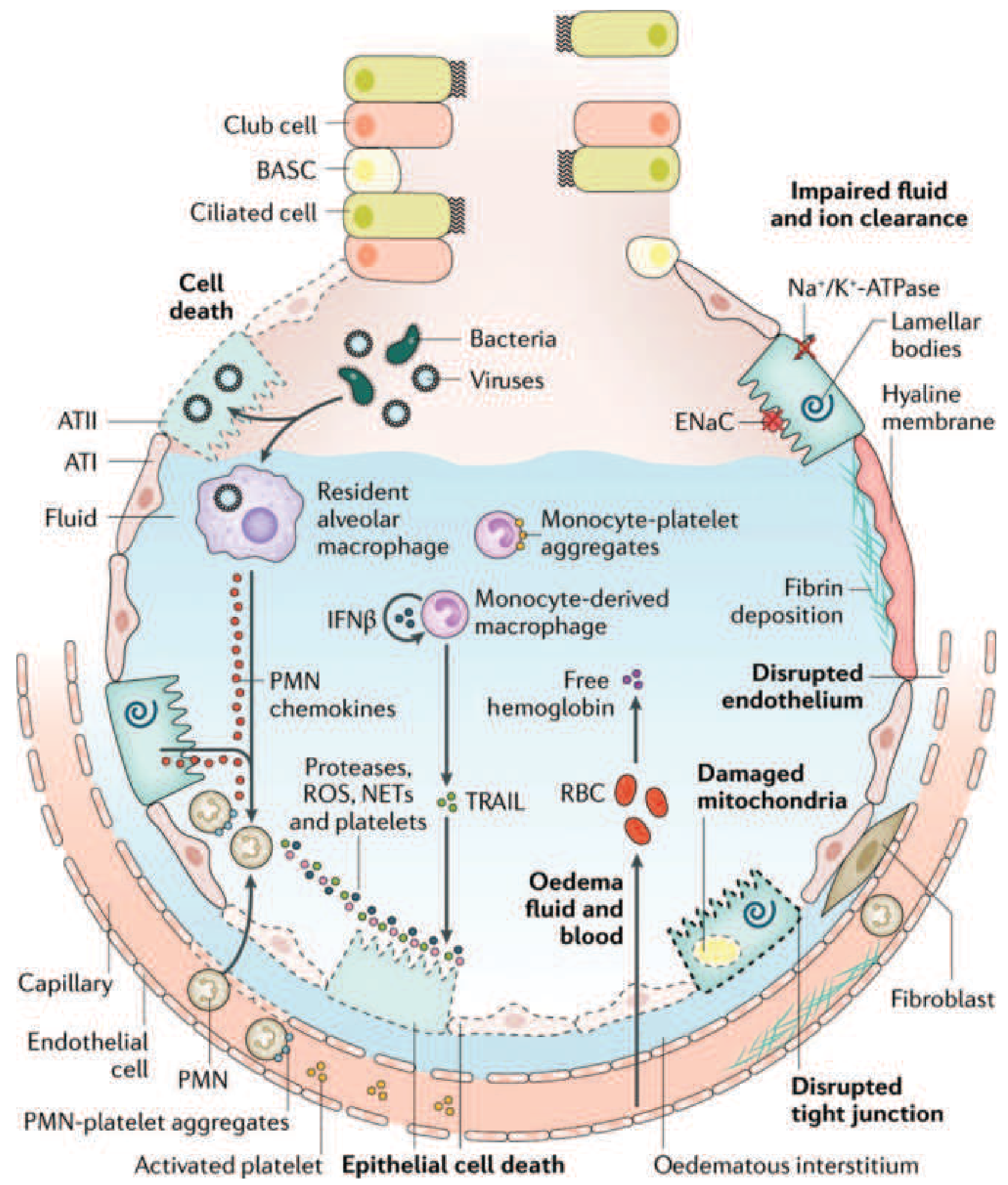
d) Consequences of Barrier Breakdown
Once the alveolar-capillary membrane is disrupted:
- Protein-rich fluid floods the alveolar space — non-cardiogenic pulmonary edema with high protein content (unlike hydrostatic edema)
- Surfactant dysfunction — both dilution and inactivation of surfactant by plasma proteins; phospholipase A2 (especially in pancreatitis) degrades surfactant directly → alveolar collapse
- Hyaline membrane formation — fibrin, cellular debris, and plasma proteins precipitate along alveolar walls — the histological hallmark
- Impaired fluid clearance — Na⁺/K⁺-ATPase and ENaC (epithelial sodium channel) on type II pneumocytes are downregulated, impairing active alveolar fluid reabsorption
- Alveolar collapse and atelectasis — loss of surfactant + edema → decreased functional residual capacity (FRC)
3. Physiological Consequences
| Mechanism | Effect |
|---|---|
| Alveolar flooding + collapse | Intrapulmonary shunt → refractory hypoxemia (V/Q mismatch) |
| Diffuse heterogeneous involvement | "Baby lung" — only ~30% of lung is ventilatable |
| Reduced lung compliance | Increased work of breathing |
| Pulmonary vasoconstriction | Hypoxic vasoconstriction + cytokine-mediated → pulmonary hypertension, right heart strain |
| Increased dead space | Microvascular thrombosis occludes perfused capillaries |
The PaO₂/FiO₂ (P/F ratio) reflects the severity: ≥200–<300 = mild; ≥100–<200 = moderate; <100 = severe (Berlin Definition).
4. The Proliferative Phase (Days 7–21)
If the patient survives the exudative phase:
- Type II pneumocytes proliferate along denuded basement membranes and differentiate into type I pneumocytes (alveolar re-epithelialization)
- Inflammatory infiltrate shifts from neutrophil-predominant → lymphocyte-predominant
- Early organization of alveolar exudates begins
- New surfactant is synthesized
5. The Fibrotic Phase (Weeks 3–4+)
A subset of patients fail to resolve and enter fibrosis:
- Alveolar exudates are replaced by interstitial and alveolar-duct fibrosis
- Intimal fibroproliferation in pulmonary microcirculation → vascular occlusion → progressive pulmonary hypertension
- Architectural destruction → emphysema-like bullae
- Results in severely reduced compliance, increased dead space, and increased pneumothorax risk
- Lung biopsy evidence of fibrosis at any stage is associated with increased mortality
6. Ventilator-Induced Lung Injury (VILI) — A Secondary Mechanism
Mechanical ventilation, while life-sustaining, can worsen ARDS via:
- Volutrauma — cyclic alveolar overdistention from excess tidal volume
- Barotrauma — excessive alveolar pressures
- Atelectrauma — repetitive collapse and reopening of alveoli
- Biotrauma — mechanical stress releases additional cytokines (IL-6, IL-8, TNF-α) from alveolar macrophages and epithelium, propagating systemic inflammation → multi-organ dysfunction syndrome (MODS)
This is why lung-protective ventilation (6 mL/kg predicted body weight, plateau pressure ≤30 cmH₂O) is the cornerstone of ARDS management. — Fishman's Pulmonary Diseases, Fig 140-3
Summary Cascade
Initiating insult (direct / indirect)
↓
TLR activation on macrophages + type I pneumocytes
↓
Chemokine release (IL-8, CXCL1) → PMN / monocyte recruitment
↓
PMN activation → elastase, MMPs, ROS, NETs
Macrophage activation → TNF-α, IL-1β, TRAIL, IFN-β
↓
Alveolar-capillary barrier disruption
(tight junction breakdown, cell death)
↓
Protein-rich edema + surfactant dysfunction + hyaline membranes
↓
Alveolar flooding → shunt → hypoxemia
Reduced compliance → increased work of breathing
↓
Exudative → Proliferative → Fibrotic phases
Sources:
- Harrison's Principles of Internal Medicine, 22E — Chapter 312, Figs 312-1, 312-3
- Murray & Nadel's Textbook of Respiratory Medicine — Chapters on ALI/ARDS pathogenesis (epithelial/endothelial injury, neutrophil mediators)
- Fishman's Pulmonary Diseases and Disorders — Chapter 140, Fig 140-3 (VALI mechanisms)
- Sabiston Textbook of Surgery — Chapter on ARDS, Berlin Definition
This is a shared conversation. Sign in to Orris to start your own chat.