Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome" AND pathogenesis
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I have everything needed for a comprehensive explanation. Let me now compile the answer.
Acute Respiratory Distress Syndrome (ARDS): Mechanism
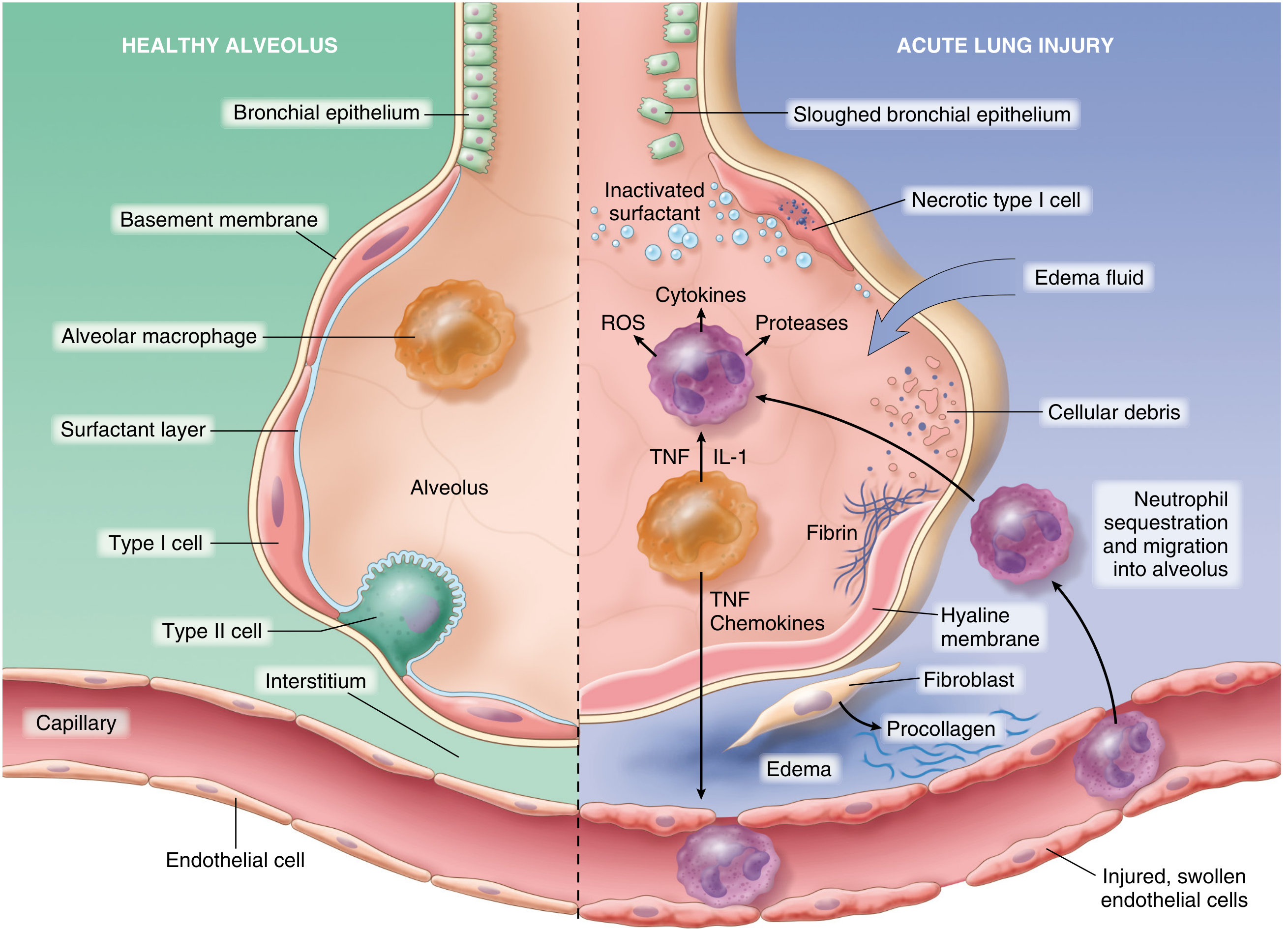
Fig. 15.3 — Normal vs. injured alveolus in ARDS. (From Robbins & Cotran Pathologic Basis of Disease)
Definition and Core Pathophysiology
ARDS is noncardiogenic pulmonary edema caused by increased alveolar-capillary membrane permeability (not elevated hydrostatic pressure). Unlike cardiogenic edema, the fluid that floods the alveoli is exudative — protein-rich — reflecting a breakdown of the barrier rather than a backup of fluid from the left heart. The result is:
- Reduced lung compliance
- Right-to-left intrapulmonary shunting → profound hypoxemia
- Increased dead-space ventilation
- Pulmonary hypertension (from hypoxic vasoconstriction, fibrin deposition, and positive-pressure ventilation)
— Murray & Nadel's Textbook of Respiratory Medicine
Initiating Events and Etiology
ARDS can be triggered by direct (pulmonary) or indirect (extrapulmonary) insults. Over 50% of cases arise from four conditions:
| Direct (Pulmonary) | Indirect (Systemic) |
|---|---|
| Pneumonia (bacterial, viral, fungal) | Sepsis |
| Aspiration of gastric contents | Severe trauma / head injury |
| Inhalation of toxic gases/smoke | Pancreatitis |
| Pulmonary contusion | Transfusion-associated lung injury (TRALI) |
| Near-drowning | Burns, DIC |
Many cases involve multiple simultaneous triggers (e.g., shock + oxygen toxicity + sepsis). The common denominator is injury to the alveolar-capillary unit.
— Robbins & Cotran Pathologic Basis of Disease
Cellular and Molecular Mechanism: Step by Step
1. Initial Injury → Endothelial and Epithelial Activation
ARDS is initiated by injury to pneumocytes (alveolar epithelial cells) and pulmonary endothelium. This can occur via two routes:
- Direct injury: Pneumocyte damage is sensed by resident alveolar macrophages, which respond by secreting TNF-α, IL-1, and other cytokines that act on neighboring endothelium.
- Indirect injury: Circulating inflammatory mediators (from sepsis, trauma, etc.) activate pulmonary endothelium directly.
Activated endothelial cells upregulate:
- Adhesion molecules (ICAM-1, E-selectin) — facilitating neutrophil attachment
- Procoagulant proteins — contributing to microvascular fibrin deposition
- Chemokines (especially IL-8) — recruiting circulating neutrophils
2. Neutrophil Sequestration and Degranulation
Neutrophils adhere to activated endothelium, migrate into the interstitium and alveolar spaces, and release:
- Proteases (elastase, matrix metalloproteinases) — directly degrade the basement membrane and extracellular matrix
- Reactive oxygen species (ROS) — oxidative damage to membranes and proteins
- Proinflammatory cytokines — amplify and perpetuate the inflammatory cascade
- Neutrophil extracellular traps (NETs) — contribute to direct tissue damage
This creates a self-amplifying feedback loop: neutrophil-mediated injury → more cytokine release → more neutrophil recruitment.
3. Alveolar-Capillary Barrier Disruption → Exudative Edema
Endothelial injury makes pulmonary capillaries leaky. Protein-rich fluid floods:
- First the interstitium
- Then the alveolar spaces
Simultaneously, type I pneumocytes (thin gas-exchange cells, 95% of alveolar surface) undergo necrosis, directly exposing the basement membrane. This disruption is the anatomic basis for the edema. The edema fluid, unlike cardiogenic edema, contains high protein concentrations and inflammatory cells.
4. Surfactant Dysfunction
Type II pneumocytes produce surfactant, which reduces alveolar surface tension and prevents collapse. In ARDS:
- Type II cells are damaged and necrotic → reduced surfactant synthesis
- Phospholipase A2 (released from inflammatory cells and leaking pancreatic enzymes) enzymatically degrades surfactant
- Protein-rich edema fluid inactivates remaining surfactant
Loss of surfactant causes alveolar collapse (atelectasis), further worsening hypoxemia and reducing compliance.
5. Hyaline Membrane Formation
The protein-rich edema fluid, cellular debris, and necrotic pneumocyte remnants coalesce and organize into hyaline membranes lining the denuded alveolar walls. This is the histologic hallmark of diffuse alveolar damage (DAD) — the pathologic correlate of ARDS.
6. Inflammation–Coagulation Crosstalk
Inflammation and coagulation are deeply interconnected in ARDS:
- TNF-α increases thrombin and fibrin formation, stimulates tissue factor expression on endothelium, and inhibits fibrinolysis
- Fibrin fragments are chemotactic for neutrophils — further amplifying recruitment
- Activated protein C (an endogenous anticoagulant) is depleted in sepsis-driven ARDS, tipping the balance toward procoagulation
- Fibrin deposition in microvasculature contributes to pulmonary hypertension and dead-space ventilation
7. Impaired Alveolar Fluid Clearance
Normally, Na⁺ channels on the apical surface of alveolar epithelial cells pump Na⁺ out of the alveolus, with water following osmotically. In ARDS:
- Hypoxia impairs expression and activity of epithelial Na⁺ channels and basolateral Na⁺/K⁺-ATPases
- This active fluid reabsorption is severely impaired, causing edema to persist and accumulate even as capillary leak is controlled
Pathologic Phases
ARDS evolves through three overlapping morphological stages:
| Phase | Timing | Pathology |
|---|---|---|
| Exudative | Days 1–7 | Hyaline membranes, alveolar flooding with protein-rich edema, neutrophilic infiltration, type I pneumocyte necrosis |
| Proliferative | Days 7–21 | Hyaline membrane resorption, fibrosis begins, type II pneumocyte proliferation to replace type I cells, decreased neutrophils |
| Fibrotic | >2–3 weeks | Pulmonary fibrosis in a subset; collagen deposition; obliteration of pulmonary capillaries |
Notably, fibroproliferation may start simultaneously with (not after) the inflammatory phase — elevated N-terminal procollagen peptide III can be detected in bronchoalveolar lavage (BAL) fluid within 24 hours of onset.
— Murray & Nadel's Textbook of Respiratory Medicine
Ventilation–Perfusion Mismatch and Hypoxemia
The pathologic lesions in ARDS are heterogeneously distributed: atelectatic, fluid-filled, and consolidated regions coexist alongside zones of nearly normal compliance and aeration. The stiff, flooded regions continue to be perfused but not ventilated, creating intrapulmonary shunt — the primary mechanism of severe hypoxemia. This also explains why ARDS hypoxemia is characteristically refractory to supplemental oxygen alone (since shunt flow bypasses gas exchange entirely regardless of FiO₂).
Genetic and Phenotypic Heterogeneity
ARDS is not a single pathologic pathway. Two distinct inflammatory subphenotypes have been identified:
- Hyperinflammatory phenotype: elevated IL-6, IL-8, soluble TNF receptor 1, lower bicarbonate; higher mortality, more vasopressor-dependent shock
- Hypo-inflammatory phenotype: lower biomarker profile, better prognosis
Genetic polymorphisms in surfactant proteins, angiopoietin pathways, and inflammatory mediators influence individual susceptibility and severity.
Summary Diagram of Key Pathways
Trigger (sepsis, pneumonia, aspiration, trauma)
│
▼
Pneumocyte/endothelial injury
│
├──► Alveolar macrophage activation → TNF-α, IL-1, IL-8
│
▼
Endothelial activation → adhesion molecules, chemokines
│
▼
Neutrophil sequestration → ROS, proteases, NETs, cytokines
│
├──► Alveolar-capillary barrier breakdown → protein-rich edema
├──► Type I pneumocyte necrosis
├──► Type II pneumocyte injury → surfactant loss/inactivation
├──► Inflammation–coagulation crosstalk → fibrin deposition
└──► Impaired Na⁺/fluid clearance
│
▼
Diffuse Alveolar Damage (DAD)
Hyaline membranes + atelectasis + V/Q mismatch
│
▼
Hypoxemic respiratory failure (ARDS)
Sources: Murray & Nadel's Textbook of Respiratory Medicine | Robbins & Cotran Pathologic Basis of Disease | Goldman-Cecil Medicine
This is a shared conversation. Sign in to Orris to start your own chat.