🩸 ROBBINS & COTRAN — CHAPTER 11
THE BLOOD VESSELS (Pathologic Basis of Disease, 10th Ed.)
═══════════════════════════════════
🔷 PART 1: VASCULAR STRUCTURE & FUNCTION (Normal)
═══════════════════════════════════
Wall Architecture
All blood vessels share the same general plan: 3 concentric layers
| Layer | Contents | Notes |
|---|
| Intima | Single EC layer + basement membrane + thin ECM | Demarcated from media by internal elastic lamina |
| Media | Smooth muscle cells (SMCs) + ECM (elastin, collagen, GAGs) | Dominant layer; thicker in arteries |
| Adventitia | Fibroblasts, nerve fibers, vasa vasorum | Outer supportive layer |
Layers are better defined in arteries than veins
Vessel Type Specializations
| Vessel Type | Key Feature | Dominant Layer | Disease Relevance |
|---|
| Elastic arteries (aorta, pulmonary) | High elastin - expand/recoil; lamellar units | Media | Loss of elastin → aneurysm |
| Muscular arteries | Circumferential SMCs; elastic laminae only at borders | Media | Atherosclerosis |
| Arterioles | Principal resistance vessels; autonomic + metabolic control | Thick media relative to lumen | Hypertension targets |
| Capillaries | Only EC + pericytes; no media | - | O₂/nutrient diffusion |
| Veins | Thin media; greater capacitance | Thin media | Stasis → thrombosis; varicosities |
Rule: Atherosclerosis → elastic + muscular arteries; Hypertension → small muscular arteries + arterioles; Vasculitis → caliber-specific
Vasa Vasorum
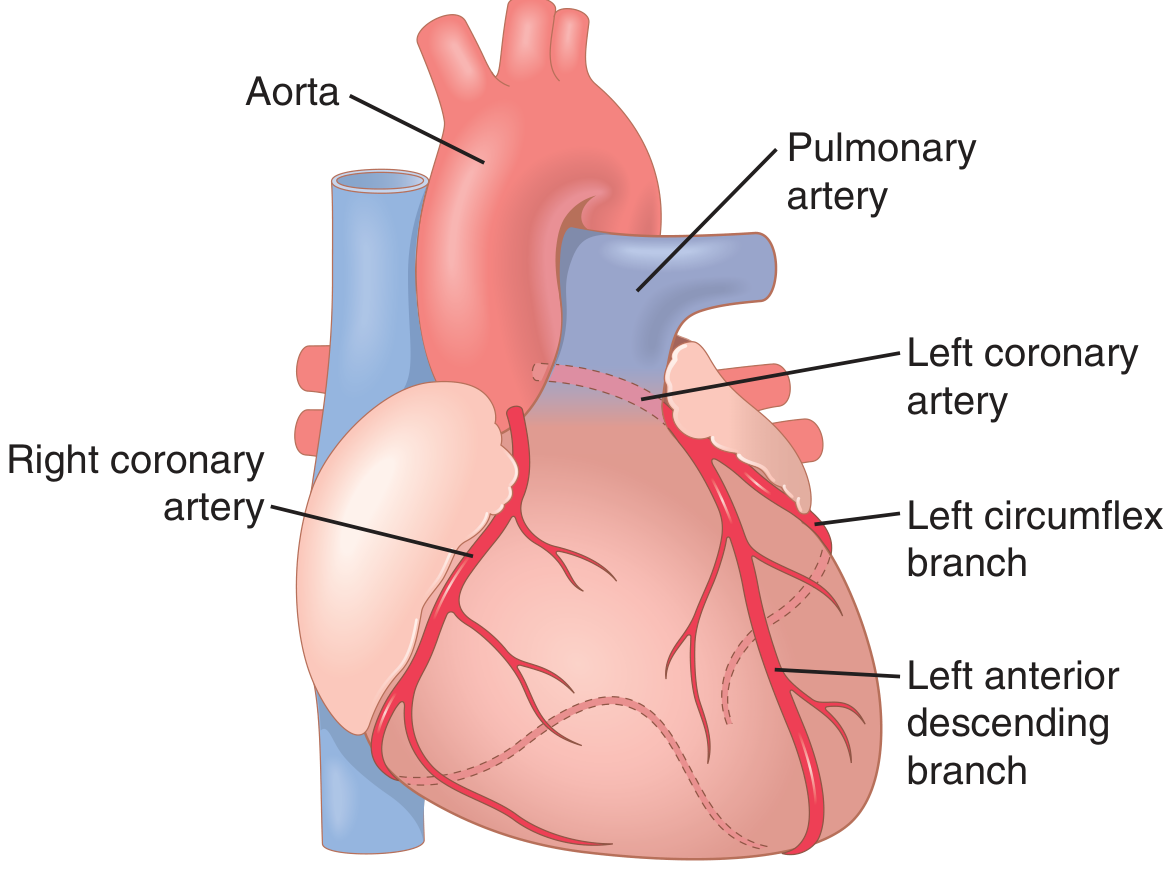
- Small vessels that supply the walls of large vessels (aorta, large arteries)
- When occluded/disrupted → medial ischemia → aneurysm or dissection
🔵 Endothelial Cell (EC) Biology
ECs are not passive conduits - they are active regulators of everything.
EC Properties & Products (Table 11.1):
| Function | Mediators |
|---|
| Permeability barrier | Tight junctions, cell–cell contacts |
| Antithrombotic | Prostacyclin (PGI₂), thrombomodulin, heparin-like molecules, plasminogen activator |
| Prothrombotic | vWF, tissue factor, plasminogen activator inhibitor (PAI) |
| ECM production | Collagen, proteoglycans |
| Vasodilation | NO, prostacyclin |
| Vasoconstriction | Endothelin, ACE |
| Inflammation regulation | IL-1, IL-6, chemokines; adhesion molecules (VCAM-1, ICAM-1, E-selectin, P-selectin) |
| Cell growth | Stimulators: PDGF, CSF, FGF; Inhibitors: heparin, TGF-β |
Endothelial Activation:
- Triggered by: cytokines (TNF, IL-1), bacterial products (LPS), turbulent/disturbed flow, lipid products (oxidized LDL), AGEs (in diabetes), viruses, complement, hypoxia
- Activated ECs: ↑ adhesion molecules, MHC molecules, cytokines, chemokines, growth factors, vasoactive/procoagulant factors
Endothelial Dysfunction:
- = Alteration of EC phenotype toward proinflammatory + prothrombogenic state
- Underlies virtually all vascular disease
🔵 Smooth Muscle Cell (SMC) Biology
| Phenotype | State | Function |
|---|
| Contractile | Normal quiescent | Vasoconstriction/dilation |
| Synthetic | Activated/injured | Proliferate, migrate, produce ECM |
- SMCs can be recruited from circulating bone marrow precursors and transition between phenotypes
- SMC proliferation and matrix synthesis → intimal thickening (fundamental response to injury)
🔴 Intimal Thickening: Stereotyped Vascular Injury Response
- The universal vessel wall response to any injury
- Sequence: EC injury/dysfunction → SMC proliferation and migration from media to intima → ECM deposition → neointima formation
- If EC is intact → neointima formation with smooth surface; SMC secretion of factors perpetuates the process
- If EC is lost → platelet adherence → thrombus → organization → fibroblast/SMC ingrowth → lesion
- This process underlies: atherosclerosis, hypertensive changes, post-angioplasty restenosis, graft failure
═══════════════════════════════════
🔷 PART 2: HYPERTENSIVE VASCULAR DISEASE
═══════════════════════════════════
Definitions
- Hypertension: systolic ≥130 mm Hg and/or diastolic ≥80 mm Hg (current guidelines)
- Affects ~30-45% of adults in developed countries
- Primary (essential) hypertension: ~95% of cases; complex polygenic + environmental
- Secondary hypertension: ~5%; identifiable cause
Mechanisms of Secondary Hypertension
| Cause | Mechanism |
|---|
| Renal artery stenosis | ↑ Renin → ↑ Ang II → vasoconstriction + Na/H₂O retention |
| Primary aldosteronism | ↑ Na/H₂O retention → ↑ blood volume |
| Pheochromocytoma | ↑↑ Catecholamines → vasoconstriction |
| Coarctation of aorta | ↓ Renal perfusion → ↑ renin; mechanical ↑ pressure above coarctation |
| Renal parenchymal disease | ↓ GFR → fluid retention; ↑ renin |
Mechanisms of Essential Hypertension
- RAAS dysregulation - abnormal salt handling, ↑ Ang II tone
- SNS hyperactivity
- Reduced nephron number (fewer nephrons from birth or injury → ↑ Na retention)
- Genetic factors - multiple polymorphisms in: Na⁺ transporters, RAAS genes, adrenergic receptors
- Environmental - high salt diet, obesity, sedentary lifestyle, stress
- Inflammation - immune cells (especially T cells) infiltrate kidney → promote Na retention
Vascular Pathology in Hypertension
1. Hyaline Arteriolosclerosis
- Seen in: benign hypertension + diabetes
- Mechanism: plasma protein leakage into walls + excessive SMC-produced matrix
- Morphology: homogeneous, pink (hyaline) thickening of arteriolar walls → narrowed lumen
- Target organs: kidneys (most important), retina, brain
2. Hyperplastic Arteriolosclerosis
- Seen in: malignant hypertension (acute severe HTN, diastolic >120 mm Hg)
- Morphology: "onion-skin" concentric laminated layers of SMCs + thickened BM → severe luminal narrowing
- Can progress to: fibrinoid necrosis → necrotizing arteriolitis → small vessel rupture → hemorrhage
- Clinical: papilledema, encephalopathy, renal failure (rapidly progressive)
═══════════════════════════════════
🔷 PART 3: ARTERIOSCLEROSIS
═══════════════════════════════════
- = Generic term for arterial wall thickening and loss of elasticity ("hardening of the arteries")
- Three patterns:
| Pattern | Vessels Affected | Key Feature |
|---|
| Atherosclerosis | Elastic + muscular arteries | Fibro-lipid plaques in intima |
| Mönckeberg medial calcific sclerosis | Medium-sized muscular arteries | Calcification of the media (does NOT narrow lumen; does NOT cause ischemia directly) |
| Arteriolosclerosis | Small arteries + arterioles | Hyaline or hyperplastic changes |
═══════════════════════════════════
🔷 PART 4: ATHEROSCLEROSIS ⭐ (Most Important)
═══════════════════════════════════
Definition
- Intimal-based lesion composed of a fibrous cap + atheromatous (lipid) core
- Affects elastic arteries (aorta, carotids, iliacs) and large/medium muscular arteries (coronaries, renals, lower extremity arteries)
- #1 cause of morbidity + mortality in developed world
Risk Factors
Major (modifiable):
- Hyperlipidemia (especially ↑ LDL, ↓ HDL)
- Hypertension
- Cigarette smoking
- Diabetes mellitus
Major (non-modifiable):
- Age (men >45 yrs; women >55 yrs post-menopause)
- Male sex
- Family history / genetics
Other (emerging):
- C-reactive protein (CRP) - marker of inflammation
- Homocysteine - causes EC injury
- Lipoprotein(a) - competes with plasminogen
- Metabolic syndrome (obesity + HTN + dyslipidemia + insulin resistance)
- Clonal hematopoiesis (TP53 mutations in blood cells → ↑ risk)
- Low socioeconomic status
Protective factors:
- HDL (reverse cholesterol transport)
- Exercise
- Moderate alcohol consumption (modest effect)
Pathogenesis: Response-to-Injury Hypothesis
Central concept: Atherosclerosis = chronic inflammatory response of the arterial wall to EC injury/dysfunction.
Step-by-step:
Step 1: ENDOTHELIAL INJURY/DYSFUNCTION
↓ (from: turbulent flow (especially at branch points),
hyperlipidemia, HTN, smoking, homocysteine, immunologic)
Step 2: LIPOPROTEIN (LDL) ACCUMULATION IN INTIMA
↓ (oxidized LDL = oxLDL is the key pathogenic form)
Step 3: MONOCYTE RECRUITMENT + MACROPHAGE DIFFERENTIATION
↓ (via adhesion molecules on activated ECs: VCAM-1, ICAM-1)
Macrophages phagocytose oxLDL via scavenger receptors
→ become FOAM CELLS (lipid-laden macrophages)
→ form "fatty streak" (earliest visible lesion)
Step 4: PLATELET ADHESION (if EC denuded)
+ T cell recruitment
↓
Step 5: SMC MIGRATION from media to intima
(driven by: PDGF from platelets/macrophages/ECs, FGF, TGF-α)
SMC phenotype switches: contractile → synthetic
↓
Step 6: ECM SYNTHESIS by SMCs
(collagen, elastin, proteoglycans → fibrous cap formation)
Step 7: LIPID ACCUMULATION
Extracellular lipid + foam cell debris → atheromatous necrotic core
Step 8: PLAQUE FORMATION = Fibrous cap + necrotic lipid core
Why branch points? Laminar flow → high shear stress → EC protective (↑ NO, ↑ antioxidants). Turbulent/disturbed flow at branch points → low shear → EC activation → pro-inflammatory → atherosclerosis.
Morphology of Atherosclerosis
Fatty Streak (earliest lesion):
- Begins as early as childhood/adolescence
- Intimal collections of foam cells (lipid-laden macrophages) + T lymphocytes
- Flat to mildly elevated; yellow
- Not yet significant clinically
- Not all fatty streaks progress to plaques
Atherosclerotic Plaque (advanced lesion):
- Fibrous cap: Dense ECM (collagen) + SMCs ± inflammatory cells
- Necrotic core: Lipid debris, foam cells, cholesterol crystals, calcification
- Shoulder regions: Most metabolically active; macrophage-rich
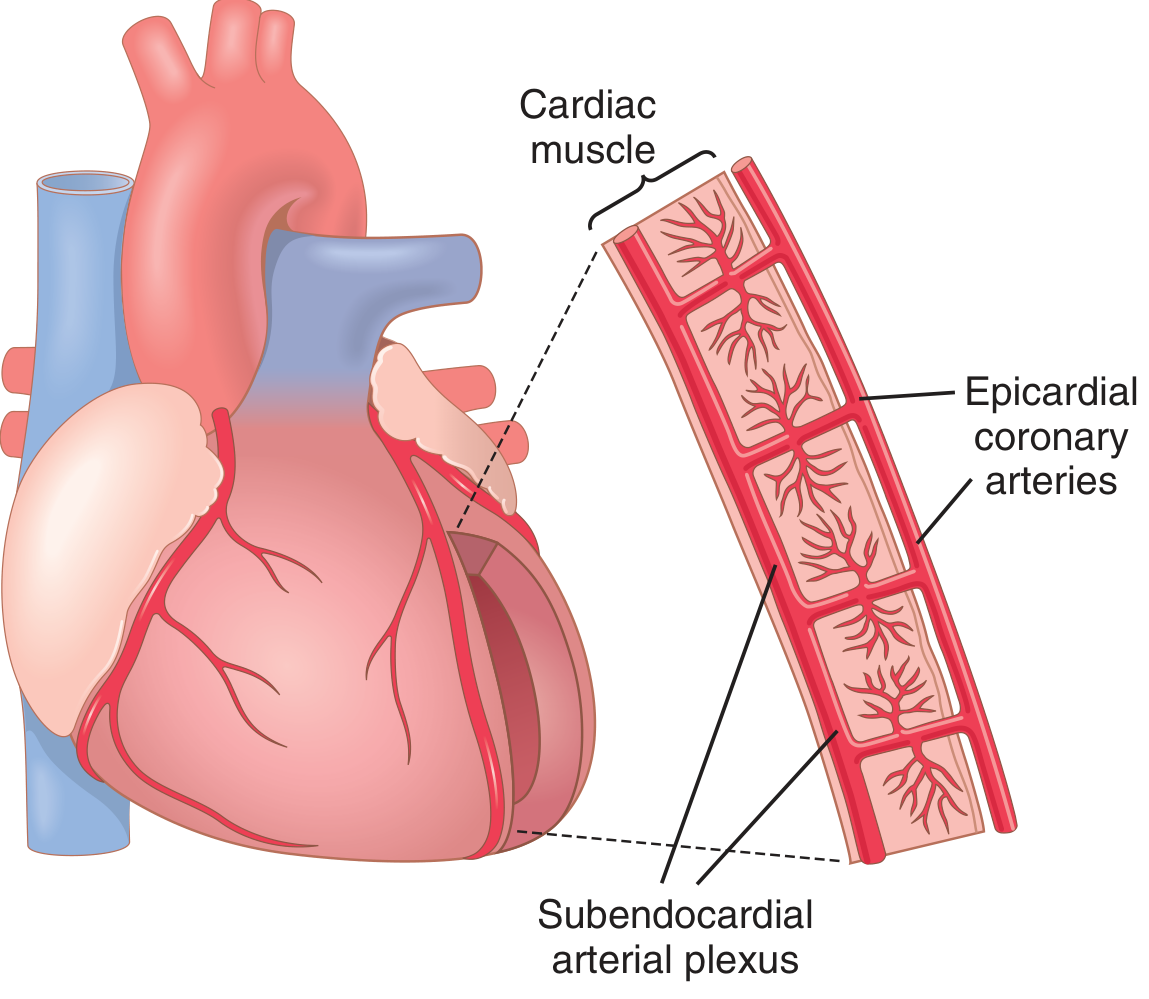
- Vasa vasorum: Can proliferate and invade the plaque base
Gross appearance:
- Creamy-yellow; patchy; irregular luminal surface
- Most severe in: abdominal aorta > coronary arteries > popliteal arteries > descending thoracic aorta > internal carotid arteries > vessels of circle of Willis
Microscopy:
- Fibrous cap: SMCs, collagen, proteoglycans
- Underlying core: foam cells, extracellular lipid, cholesterol clefts, necrotic debris
- Calcification (dystrophic) in advanced lesions
- Neovascularization (new blood vessels grow into plaque from adventitia)
Plaque Progression and Complications
Stable Plaque:
- Dense fibrous cap, minimal lipid, little inflammation
- Produces symptoms by chronic, flow-limiting stenosis → stable angina, claudication
- Lesion must narrow lumen >70% (critical stenosis) to cause ischemia at rest
Vulnerable (Unstable) Plaque:
- Thin fibrous cap, large lipid core, many macrophages and foam cells, few SMCs, more inflammation
- Prone to ACUTE PLAQUE RUPTURE (most common cause of ACS/MI/stroke)
- MMPs (from macrophages) degrade fibrous cap collagen → weakening
- Thin caps + inflammation = danger
Acute Plaque Changes (Complications):
| Complication | Mechanism | Consequence |
|---|
| Plaque rupture | Thin cap + MMP degradation | Thrombosis → ACS/MI/stroke |
| Plaque erosion | EC loss over plaque surface | Thrombosis (smaller) |
| Calcification | Dystrophic calcification in necrotic core | Plaque stiffening; visible on imaging |
| Intraplaque hemorrhage | Rupture of thin-walled plaque neovessels into core | Sudden plaque expansion → luminal narrowing |
| Thrombosis | After rupture/erosion | Occlusion → infarction |
| Embolism | Plaque debris or thrombus breaks off | Downstream ischemia |
| Aneurysm | Atherosclerosis destroys media → weakness → dilation | Rupture |
Clinical Consequences of Atherosclerosis
| Artery Affected | Clinical Syndrome |
|---|
| Coronary arteries | Angina pectoris, myocardial infarction, sudden death |
| Carotid/cerebral | TIA, ischemic stroke |
| Aorta | Aortic aneurysm, aortic dissection |
| Peripheral arteries (limbs) | Peripheral artery disease, claudication, critical limb ischemia, gangrene |
| Renal arteries | Renovascular hypertension, renal failure |
| Mesenteric arteries | Intestinal ischemia |
═══════════════════════════════════
🔷 PART 5: ANEURYSMS AND DISSECTION
═══════════════════════════════════
Definitions
| Term | Definition |
|---|
| True aneurysm | Localized dilation involving all 3 layers of intact (attenuated) arterial wall |
| False aneurysm (Pseudoaneurysm) | Defect in wall → extravascular hematoma communicates with lumen ("pulsating hematoma") |
| Dissection | Blood enters defect in wall → tunnels between medial layers (or media-adventitia) |
Types by shape:
- Saccular: Focal outpouching involving only part of the circumference
- Fusiform: Circumferential, spindle-shaped dilation of the whole vessel
Causes: Atherosclerosis (#1 overall), cystic medial degeneration (Marfan), infections (mycotic), trauma, vasculitis, syphilis
Abdominal Aortic Aneurysm (AAA)
- Location: Below the renal arteries (infrarenal) - most common
- Size threshold for rupture risk: >5 cm diameter
- Cause: Atherosclerosis (>95%) → medial destruction by lipid/inflammation + ↓ elastin/collagen synthesis
- Risk factors: Age >60, male sex, smoking (major risk!), family history, HTN
- Morphology: Fusiform dilation of infrarenal aorta; luminal mural thrombus (atherosclerotic)
- Complications:
- Rupture (catastrophic - mortality 50-80% even with surgery)
- Thrombosis → downstream embolism/ischemia
- Compression of adjacent structures (ureters, vertebrae, duodenum)
- Atherosclerotic embolism
- Screening: Ultrasound for men aged 65-75 who ever smoked
- Variants:
- Inflammatory AAA (5-10%): Dense peri-aortic fibrosis + inflammatory infiltrate; some associated with IgG4-related disease
- Mycotic AAA: Bacterial/fungal seeding of aortic wall (Pseudomonas, Salmonella); hematogenous or from adjacent infection
Thoracic Aortic Aneurysm (TAA)
- Cause: Cystic medial degeneration >> atherosclerosis
- Associations:
- Marfan syndrome (FBN1 mutation → ↓ fibrillin-1 → ↑ TGF-β signaling → elastin breakdown)
- Bicuspid aortic valve (associated structural ECM weakness of aortic root)
- Hypertension (major accelerating factor)
- Tertiary syphilis → endarteritis obliterans of vasa vasorum → medial ischemia → ascending aorta aneurysm (classically involves aortic root → aortic valve regurgitation)
- Morphology: Fusiform, usually ascending aorta; cystic medial change = loss of elastic fibers + SMC dropout + mucoid/cystic spaces
- Complications:
- Aortic valve regurgitation (if involves aortic root)
- Superior vena cava syndrome
- Dysphagia, hoarseness, cough (compression of adjacent structures)
- Rupture
- Dissection
Aortic Dissection
- Blood splits through the media (not just intima) creating a false lumen
- NOT the same as aneurysm - dissection can occur without prior aneurysm (though often coexists)
- Can also arise from rupture of vasa vasorum within media
Causes/Risk factors:
- Hypertension (#1 precipitant - present in ~75%)
- Cystic medial degeneration (Marfan syndrome, bicuspid aortic valve, Ehlers-Danlos)
- Age 40-60 in HTN patients; younger in Marfan/connective tissue disorders
- Pregnancy (especially 3rd trimester - hormonal changes weaken aortic wall)
- Rare: iatrogenic (cardiac catheterization), trauma, cocaine use
Classification (DeBakey/Stanford):
| Type | Extent | Risk | Management |
|---|
| Type A (proximal) | Involves ascending aorta (with or without arch) | Highest mortality; ~1-2% per hour acutely | Surgical emergency |
| Type B (distal) | Descending aorta only (distal to left subclavian) | Lower immediate risk | Medical management (BP control); surgery if complications |
Morphology:
- Entry tear usually in ascending aorta within 10 cm of aortic valve, or just distal to left subclavian origin
- False lumen (between inner 2/3 and outer 1/3 of media) fills with clotted blood
- Can propagate proximally and/or distally
- May re-enter lumen downstream (double-barrel aorta) or rupture outward
Complications of Type A:
- Hemopericardium → cardiac tamponade (most deadly acute complication)
- Aortic valve insufficiency (disruption of leaflet support)
- MI (occlusion of coronary ostia)
- Stroke (occlusion of carotid ostia)
- Rupture into mediastinum/pleural cavity
Complications of Type B:
- Mesenteric/renal ischemia
- Lower limb ischemia
- Rupture (less common acutely)
═══════════════════════════════════
🔷 PART 6: VASCULITIS
═══════════════════════════════════
Definition & Overview
- = Inflammation of vessel walls
- Manifestations depend on vascular bed involved
- Systemic: fever, myalgias, arthralgias, malaise + organ-specific features
Two Pathogenic Mechanisms:
- Direct infectious invasion of vessel wall
- Immune-mediated (most common):
- Immune complex deposition
- ANCA (antineutrophil cytoplasmic antibody)
- Anti-EC antibodies + autoreactive T cells
- Anti-GBM antibodies
⚠️ Must distinguish infectious vs. immune → immunosuppression helps immune, harms infectious
Infectious Vasculitis
- Agents: Pseudomonas, Aspergillus, Mucor most common
- Via: local tissue spread from adjacent infection, or hematogenous
- Consequences: mycotic aneurysm formation, thrombosis → downstream infarction
- Example: bacterial meningitis → inflammation-induced thrombosis of meningeal vessels → brain infarction
Noninfectious Vasculitis - Classification by Vessel Size
> 25 primary forms recognized. Key ones:
🔴 LARGE VESSEL VASCULITIS
1. Giant Cell (Temporal) Arteritis
- Most common vasculitis in developed countries
- Age: Almost exclusively in people >50 years (peak 70-80s)
- Sex: Female >> male (2:1)
- Vessels: Temporal artery (classic), also ophthalmic, vertebral, internal carotid; less often aorta and its major branches
Pathogenesis:
- Granulomatous inflammation triggered by unknown antigen in vessel wall
- Activated CD4⁺ T cells + macrophages → granuloma with giant cells
- IL-6 markedly elevated (correlates with disease activity; basis for IL-6 blockade therapy)
Morphology:
- Granulomatous inflammation of inner media → giant cells (Langhans and foreign-body type) clustered around fragmented internal elastic lamina
- Intimal thickening → luminal narrowing
- Skip lesions (not uniform along vessel)
- Late stage: fibrotic healing → "pipestem" artery
Clinical Features:
- Temporal headache (most classic)
- Tenderness and nodularity/thickening of temporal artery; absent pulse
- Jaw claudication (pain on chewing) - from ischemia of masseter
- Sudden irreversible blindness - most feared complication (from ophthalmic artery involvement)
- Fever, malaise, weight loss, elevated ESR/CRP
- Often associated with polymyalgia rheumatica (shoulder/hip girdle pain and stiffness in ~50% of GCA patients)
- ↑ IL-6
Diagnosis: Temporal artery biopsy (skip lesions → long biopsy required)
Treatment: High-dose corticosteroids → immediate initiation to prevent blindness; tocilizumab (anti-IL-6R) for refractory/relapsing disease
2. Takayasu Arteritis ("Pulseless Disease")
- Age: <50 years (usually 20s-40s)
- Sex: Female >> male (9:1)
- Geographic predilection: Asia, Middle East, Latin America
- Vessels: Aorta + great branches (subclavian, renal, carotid, mesenteric, coronary)
Pathogenesis: Granulomatous inflammation; cell-mediated immunity; NK cell + γδ T cell involved
Morphology:
- Granulomatous adventitial inflammation → progresses to severe fibrous thickening of aortic wall and branch ostia
- Gross: irregular intimal plaques; tree bark-like intima
- Late: progressive luminal narrowing of aortic branches
Clinical Features:
- Absent pulses in upper extremities (subclavian stenosis) → pulse difference between arms
- BP differential between arms >10 mm Hg
- Ocular disturbances, retinal hemorrhages (carotid/ophthalmic stenosis)
- Neurological symptoms (carotid stenosis)
- Hypertension (renal artery stenosis)
- Aortic regurgitation (if aortic root involved)
- Constitutional symptoms: fever, fatigue, weight loss, elevated ESR
Treatment: Corticosteroids; methotrexate; TNF inhibitors for refractory
🟠 MEDIUM VESSEL VASCULITIS
3. Polyarteritis Nodosa (PAN)
- Affects medium-sized muscular arteries (renal, hepatic, coronary, mesenteric)
- Does NOT involve: pulmonary vessels, glomerular vessels
- No ANCA association
- Hepatitis B association in ~30% of cases (immune complex deposition)
- Can occur at any age; more common in young adults
Pathogenesis: Immune complex deposition in vessel walls → complement activation → neutrophil recruitment → necrotizing inflammation
Morphology:
- Segmental (not continuous), necrotizing inflammation affecting entire vessel wall
- Fibrinoid necrosis of wall + transmural inflammatory infiltrate
- Skip lesions (normal segments between diseased segments)
- Acute: fibrinoid necrosis + neutrophils
- Healing: fibrosis, obliteration, aneurysmal dilation at branch points (classic "beaded" pattern on angiography)
Clinical Features:
- Fever, weight loss, malaise
- Hypertension (renal involvement)
- Abdominal pain (mesenteric ischemia)
- Peripheral neuropathy (vasa nervorum involvement) - very common
- Myalgias, arthralgias
- Skin nodules, livedo reticularis, ulcers
- Renal involvement → hematuria, proteinuria, renal failure
Diagnosis: Angiography (microaneurysms at branch points), biopsy
Treatment: Steroids ± cyclophosphamide; antiviral therapy if HBV-associated
4. Kawasaki Disease (Mucocutaneous Lymph Node Syndrome)
- Age: Children <5 years (peak 6-24 months); rare in adults
- Ethnicity: Originally described in Japan; higher incidence in Asian populations
- Cause: Unknown; likely infectious trigger + immune dysregulation
Pathogenesis: Immune complex-mediated and T cell-mediated; IL-1 may be important (basis for anti-IL-1 therapy)
Morphology:
- Acute: Necrotizing arteritis of coronary arteries → coronary artery aneurysms (in 15-25% untreated)
- Resembles PAN histologically in acute phase
Clinical Features (diagnostic criteria - 5 of 6):
- Fever ≥5 days
- Bilateral conjunctival injection (non-exudative)
- Oral mucosa changes (strawberry tongue, lip cracking/erythema, pharyngeal erythema)
- Rash (polymorphous exanthem)
- Extremity changes (edema, erythema of palms/soles → desquamation)
- Cervical lymphadenopathy (often unilateral, ≥1.5 cm)
Most feared complication: Coronary artery aneurysms → thrombosis → MI (leading cause of acquired heart disease in children in developed countries)
Treatment: IV immunoglobulin (IVIG) + aspirin (reduces coronary aneurysm risk from 25% to <5%)
🟡 SMALL VESSEL VASCULITIS
ANCA-Associated Vasculitides (AAV)
ANCA = antineutrophil cytoplasmic antibodies
- c-ANCA (PR3-ANCA): anti-proteinase-3; pattern = cytoplasmic
- p-ANCA (MPO-ANCA): anti-myeloperoxidase; pattern = perinuclear
Mechanism: ANCA activates primed neutrophils → neutrophil degranulation against vessel walls → vascular damage without immune complex deposition ("pauci-immune")
5. Granulomatosis with Polyangiitis (GPA) (formerly Wegener)
- c-ANCA (PR3-ANCA) positive in ~90%
- Triad: upper respiratory tract + lower respiratory tract + kidneys
- Classic triad: Necrotizing granulomas of upper/lower RT + necrotizing vasculitis of small/medium vessels + focal necrotizing glomerulonephritis
Clinical Features:
- Saddle-nose deformity (nasal cartilage destruction)
- Chronic sinusitis, otitis media, epistaxis
- Oral/nasal ulcers with tissue destruction
- Pulmonary: nodules/cavities, hemoptysis, dyspnea
- Renal: rapidly progressive glomerulonephritis (RPGN) → hematuria, proteinuria, renal failure
- Eyes: conjunctivitis, proptosis, orbital pseudotumor (granuloma)
Morphology: Necrotizing granulomas (with giant cells) + necrotizing vasculitis of vessels
Treatment: Rituximab (anti-CD20) or cyclophosphamide + glucocorticoids
6. Eosinophilic Granulomatosis with Polyangiitis (EGPA) (formerly Churg-Strauss)
- p-ANCA (MPO-ANCA) positive in ~40%
- Classic triad: Asthma + eosinophilia + systemic vasculitis
- Mainly upper/lower respiratory tract involvement initially → then systemic
Clinical Features:
- Asthma (almost always present - hallmark)
- Peripheral + tissue eosinophilia
- Allergic rhinitis, nasal polyps
- Cardiac involvement (eosinophilic myocarditis/endocarditis) - major cause of death
- Peripheral neuropathy (mononeuritis multiplex)
- Skin nodules, purpura
Morphology: Granulomatous inflammation + eosinophilic infiltrate in vessel walls
Treatment: Steroids; cyclophosphamide for severe cases; mepolizumab (anti-IL-5)
7. Microscopic Polyangiitis (MPA)
- p-ANCA (MPO-ANCA) positive in ~70%
- Affects: small vessels (capillaries, venules, arterioles); also medium arteries sometimes
- No granulomas (unlike GPA and EGPA)
- Pauci-immune (no/minimal immune complex deposition)
Clinical Features:
- Necrotizing glomerulonephritis (RPGN) - most common cause of death
- Pulmonary capillaritis → diffuse alveolar hemorrhage
- Skin purpura
- Peripheral neuropathy
- Abdominal pain, GI bleeding
Treatment: Rituximab or cyclophosphamide + steroids; plasma exchange for severe renal/pulmonary disease
Immune Complex-Associated Small Vessel Vasculitis
8. IgA Vasculitis (Henoch-Schönlein Purpura/HSP)
- Most common vasculitis in children
- IgA immune complex deposition in small vessel walls
- Classic tetrad:
- Purpura (non-thrombocytopenic; palpable; lower extremities/buttocks)
- Arthritis/arthralgias
- Abdominal pain/GI bleeding (bowel ischemia)
- Renal disease (IgA nephropathy-like; hematuria ± nephrotic syndrome)
- Often follows upper respiratory infection (IgA-triggering)
- Treatment: supportive; steroids for severe cases
9. Cryoglobulinemic Vasculitis
- Immune complex deposition with cryoglobulins (immunoglobulins that precipitate at cold temperatures)
- Most commonly: Hepatitis C infection (mixed cryoglobulinemia type II/III)
- Clinical: purpura, arthralgia, weakness, peripheral neuropathy, glomerulonephritis
- Treatment: treat underlying HCV infection; steroids/rituximab for severe cases
Anti-GBM Antibody Disease (Goodpasture Syndrome)
- Anti-GBM antibodies cross-react with pulmonary alveolar BM
- Glomerulonephritis (RPGN) + pulmonary hemorrhage
- Treated with plasma exchange + immunosuppression
═══════════════════════════════════
🔷 PART 7: DISORDERS OF BLOOD VESSEL HYPERREACTIVITY
═══════════════════════════════════
Raynaud Phenomenon
- Exaggerated vasoconstriction of arteries/arterioles in response to cold or emotion
- Affects: fingers/toes most commonly; also nose, earlobes, lips
- Classic color change: Red (proximal vasodilation) → White (vasoconstriction) → Blue (distal cyanosis) ("red-white-blue" from proximal to distal)
| Primary Raynaud | Secondary Raynaud |
|---|
| Prevalence | 3-5% general population | Less common |
| Demographics | Young females; bilateral/symmetric | Any age; asymmetric |
| Cause | Intrinsic SMC hyperreactivity (no structural abnormality) | Underlying disease: SLE, scleroderma, Buerger disease, atherosclerosis |
| Progression | Generally non-progressive; benign | Progressive |
| Complications | Rare; skin atrophy; gangrene very rare | More common; ulceration, gangrene |
| Structural wall change | Absent (except late intimal thickening in long-standing) | Present |
⚠️ Raynaud phenomenon may be the FIRST manifestation of immune-mediated vasculitis. New onset → evaluate for underlying disease. ~10% will eventually manifest a systemic disorder.
Myocardial Vessel Vasospasm
- Excessive constriction of coronary arteries/arterioles
- Can produce variant (Prinzmetal) angina - rest pain, ST elevation, no fixed stenosis
- Triggers: cold, stress, cocaine, alpha-adrenergic agonists
- More common in arteries with underlying atherosclerosis (even mild plaques)
- Can precipitate MI and sudden death
- Treatment: calcium channel blockers + nitrates (avoid beta-blockers)
═══════════════════════════════════
🔷 PART 8: VEINS AND LYMPHATICS
═══════════════════════════════════
Varicose Veins
- = Abnormally dilated, tortuous veins caused by chronically increased intraluminal pressure
- Most common sites: lower extremity superficial veins (great + small saphenous system)
- Prevalence: ~15% of adults
Predisposing factors:
- Prolonged standing (occupation - surgeons, teachers, nurses, etc.)
- Pregnancy (↑ blood volume + venous pressure + hormonal relaxation of vein walls)
- Obesity
- Hereditary weakness of venous walls
- Prior DVT (damages valves → valve incompetence → secondary varicosities)
Pathogenesis:
- Venous valve incompetence (incompetent valves) → blood refluxes downward → ↑ pressure → venous dilation → further valve incompetence (vicious cycle)
Morphology:
- Tortuous, dilated, elongated veins
- Irregular wall thickening (hypertrophied smooth muscle in areas of dilation; atrophied in others)
- Intimal fibrosis
- Calcified thrombi ("phleboliths")
Clinical Consequences:
- Stasis → impaired venous drainage → chronic venous insufficiency
- Varicose ulcers (especially medial malleolus) - from tissue hypoxia/ischemia
- Poor healing of wounds
- Thrombosis of superficial varices (less dangerous than DVT)
- Variceal hemorrhage (if significant trauma)
- Cosmetic concerns
Varices at other sites:
- Esophageal varices: Portal hypertension → portosystemic shunting through coronary vein → gastroesophageal veins; catastrophic hemorrhage risk
- Hemorrhoidal veins: Engorgement of the hemorrhoidal plexus; symptomatic hemorrhoids
- Varicocele: Dilation of pampiniform plexus of spermatic vein (left > right); associated with male infertility
Thrombophlebitis and Phlebothrombosis
- Thrombophlebitis: Venous thrombosis in setting of primary inflammation (infectious or sterile)
- Phlebothrombosis: Venous thrombosis without primary inflammation (Virchow's triad)
- In practice, these terms are used interchangeably because they co-exist
Sites:
- Deep veins of legs (90% of DVT): calf, popliteal, femoral, iliac
- Mesenteric veins (hypercoagulable states)
- Hepatic veins (Budd-Chiari syndrome)
- Dural venous sinuses (intracranial thrombosis)
- Portal vein
Risk factors (Virchow's Triad):
- Stasis - immobility, cardiac failure, pregnancy, varicosities
- Hypercoagulability - factor V Leiden, antiphospholipid syndrome, cancer, OCP, nephrotic syndrome
- Endothelial injury - trauma, surgery, indwelling catheters
Migratory thrombophlebitis (Trousseau Syndrome):
- Recurrent venous thromboses at unusual/migratory sites
- Association with occult visceral carcinoma (pancreatic, lung, gastric, colon)
- Tumor-secreted procoagulant factors
Clinical consequences of DVT:
- Pulmonary embolism (major cause of sudden death)
- Post-thrombotic syndrome (valve damage → chronic venous insufficiency, ulcers)
- Phlegmasia cerulea dolens (massive proximal DVT → entire limb cyanosis/pain)
Superior Vena Cava (SVC) Syndrome
- Cause: Compression or invasion of SVC
- Most common cause: Bronchogenic carcinoma (right upper lobe) or lymphoma
- Clinical:
- Progressive facial/neck/arm swelling (pitting edema) and cyanosis
- Edema of upper extremities and neck ("collar of stokes")
- Dilated superficial veins on chest wall (collateral drainage)
- Headache, visual disturbances (↑ cerebral venous pressure)
- Potentially fatal if cerebral edema develops
Inferior Vena Cava (IVC) Syndrome
- Cause: Compression by adjacent tumor, hepatic abscess, retroperitoneal fibrosis; thrombosis
- Common associations: hepatocellular carcinoma (invades IVC), renal cell carcinoma (tumor thrombus extends into IVC)
- Clinical:
- Leg edema
- Distension of superficial collateral vessels of the abdominal wall
- Massive proteinuria (if renal veins obstructed)
Lymphangitis and Lymphedema
Lymphangitis:
- Acute: Bacterial infection → spreads along lymphatics → red streaks under skin ("blood poisoning" - misnomer); most commonly group A Streptococcus
- Can lead to bacteremia/septicemia
- Regional lymph nodes enlarge and become painful (reactive lymphadenopathy)
Lymphedema:
- Accumulation of lymph due to lymphatic obstruction → protein-rich edema
- Primary (congenital):
- Milroy disease: congenital hereditary lymphedema (lower extremities; VEGFR3 mutation)
- Simple primary lymphedema (idiopathic onset in 2nd-3rd decade)
- Secondary (most common):
- Lymph node dissection (post-mastectomy → arm lymphedema "brawny arm")
- Radiation-induced lymphatic fibrosis
- Filariasis (Wuchereria bancrofti) - most common cause worldwide → massive scrotal/lower extremity lymphedema = elephantiasis
- Tumor obstruction of lymphatics
- Recurrent infections
- Complications of chronic lymphedema:
- Protein-rich edema → fibrosis → "brawny" (non-pitting) induration
- Risk of lymphangiosarcoma (Stewart-Treves syndrome after mastectomy)
═══════════════════════════════════
🔷 PART 9: VASCULAR TUMORS
═══════════════════════════════════
Classification
| Grade | Examples |
|---|
| Benign/Tumor-like | Vascular ectasias, hemangiomas, lymphangiomas, glomus tumor, bacillary angiomatosis |
| Intermediate (borderline) | Kaposi sarcoma, hemangioendothelioma |
| Malignant | Angiosarcoma |
Benign Tumors and Tumor-like Conditions
Vascular Ectasias (Telangiectasias)
- = Localized permanent dilation of pre-existing vessels (NOT true tumors)
- Types:
- Nevus flammeus (port-wine stain): Flat, purplish lesion on face/neck; irregular superficial vessels; grows with child; does NOT involute; may be associated with Sturge-Weber syndrome (if V1 distribution → leptomeningeal angioma → epilepsy/intellectual disability)
- Spider telangiectasia (spider angioma): Central feeding arteriole with radiating small vessels; seen in cirrhosis (↑ estrogen) and pregnancy; blanches on pressure; refills from center
- Hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu): AD; ENG/ACVRL1 mutations (TGF-β pathway); telangiectasias in mucous membranes, GI tract, skin, lungs → recurrent epistaxis, GI bleeding, AV malformations
Hemangiomas
- Most common benign vascular tumors; occur in all ages
- Most frequently in skin and soft tissues
| Type | Features |
|---|
| Capillary hemangioma | Most common; small vessels; lobulated; skin, mucous membranes, liver, spleen, kidneys |
| Strawberry hemangioma (infantile) | Most common tumor of infancy; grows rapidly, then INVOLUTES by age 5-8 (unlike nevus flammeus) |
| Cavernous hemangioma | Large, dilated vascular channels; deeper tissues; brain (cerebellum in VHL), liver |
| Pyogenic granuloma | Rapidly growing polypoid capillary hemangioma; skin/mucous membranes after minor trauma; bleeds easily; mistaken for malignancy |
VHL association: Cerebellar hemangioblastoma = classic finding in von Hippel-Lindau syndrome
Lymphangiomas
- Benign tumors of lymphatic vessels
- Simple (capillary) lymphangioma: Occur in head/neck of children; small vessels lined by EC
- Cavernous lymphangioma (cystic hygroma): Dilated lymphatic spaces; most common in neck; associated with Turner syndrome (45,X) and Down syndrome
Glomus Tumor (Glomangioma)
- Benign, extremely painful tumor arising from modified SMCs of glomus bodies (arteriovenous shunts in skin)
- Most common in subungual region of fingers (under fingernails)
- Also: distal extremities
- Morphology: small; nests/organoids of round uniform cells surrounding vascular spaces
- Treatment: surgical excision → curative; exquisitely painful on pressure
Bacillary Angiomatosis
- Vascular proliferation caused by Bartonella henselae or B. quintana infection
- Seen almost exclusively in HIV/AIDS patients and other immunocompromised
- NOT a true neoplasm - it is an infectious vascular proliferation
- Morphology: looks like Kaposi sarcoma; lobular proliferation of plump (epithelioid) ECs; neutrophilic infiltrate; clumps of bacilli (Warthin-Starry silver stain)
- Treatment: Erythromycin (antibiotic)
Intermediate (Borderline) Tumors
Kaposi Sarcoma (KS)
- Vascular tumor caused by HHV-8 (Human Herpesvirus-8/Kaposi Sarcoma Herpesvirus)
- Exists in several clinical forms
| Form | Population | Behavior |
|---|
| Classic (sporadic) KS | Elderly men of Eastern European/Mediterranean descent; not immunocompromised | Indolent; lower limb skin lesions; internal organ involvement rare |
| Lymphadenopathic (African) KS | Young Africans (children); endemic areas | Aggressive; lymph node involvement |
| Transplant-associated KS | Organ transplant recipients (calcineurin inhibitors suppress T cells → allow HHV-8) | Regresses if immunosuppression reduced |
| AIDS-related KS | HIV+ patients (CD4 <200); gay men most affected | Aggressive; widespread; visceral involvement |
Pathogenesis:
- HHV-8 infects ECs → immortalization
- CD4 T-cell immunosuppression → allows HHV-8 replication
- HHV-8 proteins: LANA-1 (inhibits p53 + RB), vIL-6 (viral homologue), vCCL-2/3 (viral chemokines), vCyclin (CDK6 activator → cell proliferation)
- Not a true cancer - it is a reactive/proliferative lesion driven by HHV-8
Morphology:
- Patch stage: Dilated, irregular, angular vascular channels in dermis; few atypical ECs; barely visible
- Plaque stage: More evident; confluent vascular channels lined by spindle cells
- Nodular stage: Cellular; sheets of spindle-shaped ECs; slit-like vascular channels; extravasated RBCs (hemosiderin deposits); mitoses
Clinical:
- Skin: painless purple/brown macules → nodules; lower extremities → disseminates
- AIDS-KS: oral mucosa, GI tract, lungs, lymph nodes → severe complications
- Treatment: for AIDS-KS → HAART (antiretroviral therapy) can induce regression; localized → radiation; systemic → chemotherapy (liposomal doxorubicin, paclitaxel)
Hemangioendothelioma
- Intermediate malignant potential (between hemangioma and angiosarcoma)
- Epithelioid hemangioendothelioma: liver, lung; short spindle to rounded EC tumor cells; variable behavior
- Treatment: wide local excision; some require chemo
Malignant Tumors
Angiosarcoma (Hemangiosarcoma)
- Malignant tumor of vascular endothelial cells
- Can arise in any tissue but most common in: skin, soft tissue, breast, liver
Associated conditions/risk factors:
- Post-mastectomy lymphedema → cutaneous angiosarcoma of arm (Stewart-Treves syndrome)
- Thorotrast (old radiologic contrast - thorium dioxide) → hepatic angiosarcoma
- Vinyl chloride → hepatic angiosarcoma
- Arsenic → hepatic angiosarcoma
- Chronic lymphedema → cutaneous angiosarcoma
- Radiation-associated (secondary to prior radiotherapy)
Morphology:
- Poorly defined margins; gray-white lesions ± hemorrhage
- Histology: variable - well-differentiated (distinct vascular channels with atypical ECs) to poorly differentiated (solid sheets, few vascular spaces)
- IHC markers: CD31, CD34, ERG (endothelial markers)
- Aggressive: local invasion + hematogenous spread
Clinical: Poor prognosis; 5-year survival ~30% for cutaneous; worse for visceral
═══════════════════════════════════
🔷 PART 10: PATHOLOGY OF VASCULAR INTERVENTION
═══════════════════════════════════
Endovascular Stenting (Angioplasty + Stent)
Process:
- Balloon catheter inflates → stretches/compresses plaque → opens lumen
- Stent (metal mesh) deployed → mechanically maintains luminal patency
Complications and Pathology:
1. Acute:
- Elastic recoil of vessel wall immediately after balloon deflation
- Stent prevents this mechanical recoil
2. Restenosis (occurs in 30-50% of bare-metal stents within 6-12 months):
- Mechanism: Vascular injury during angioplasty → intimal response (smooth muscle proliferation + ECM) → neointimal hyperplasia → luminal renarrowing
- Not atherosclerosis re-accumulating; it is a wound-healing response
Drug-Eluting Stents (DES):
- Coated with anti-proliferative drugs (paclitaxel, sirolimus/rapamycin, everolimus)
- Drugs inhibit SMC proliferation → markedly reduce restenosis (<10% at 1 year)
- BUT: DES delay endothelialization → risk of late stent thrombosis (months to years) if antiplatelet therapy stopped → dual antiplatelet therapy (aspirin + P2Y12 inhibitor) required for 6-12 months minimum
3. In-stent thrombosis:
- Bare metal: early (hours to days) from incomplete stent apposition or platelet activation
- Drug-eluting: late/very late (months to years) due to impaired re-endothelialization
Vascular Replacement (Bypass Grafts)
Types of grafts:
- Autologous vein grafts (saphenous vein, internal mammary artery) - best long-term patency
- Prosthetic grafts (Dacron, PTFE/Gore-Tex) - used when autologous vessels unavailable
Pathology of Graft Failure:
| Time | Cause | Mechanism |
|---|
| Early (<1 month) | Acute thrombosis | Technical error, poor flow, hypercoagulable state |
| Intermediate (1 month - 2 years) | Intimal hyperplasia | SMC proliferation at anastomosis sites |
| Late (>2 years) | Atherosclerosis of graft | Progressive occlusive disease |
Autologous vein graft arterialization:
- When vein placed in arterial position → exposed to higher pressures and flow
- Develops: intimal hyperplasia, medial hypertrophy → eventually becomes "arterialized"
- Long-term: atherosclerosis develops in graft, especially at anastomoses
Prosthetic graft complications:
- Thrombosis (especially small-diameter grafts)
- Graft infection → often requires graft removal (life-threatening)
- Anastomotic pseudoaneurysm (at suture line between graft and natural artery)
- Graft-to-bowel fistula (aorto-enteric; rare but fatal)
═══════════════════════════════════
📋 MASTER SUMMARY TABLE
═══════════════════════════════════
| Topic | Key Points |
|---|
| EC function | Antithrombotic, antiinflammatory, vasodilatory at rest; switches to prothrombotic, proinflammatory when activated/injured |
| Hyaline arteriolosclerosis | Benign HTN + DM; pink homogeneous walls; kidney target |
| Hyperplastic arteriolosclerosis | Malignant HTN; onion-skin; fibrinoid necrosis |
| Atherosclerosis pathogenesis | EC dysfunction → LDL oxidation → macrophage foam cells → SMC migration → fibrous cap + necrotic core |
| Vulnerable plaque | Thin cap, large lipid core, many macrophages, MMP-mediated rupture |
| Critical stenosis | >70% narrowing needed for rest-ischemia |
| AAA | Below renal arteries; >5cm → risk of rupture; smoking/aging/male |
| TAA | Cystic medial degeneration; Marfan; bicuspid AV; syphilis (ascending) |
| Dissection Type A | Ascending aorta; tamponade/AR/MI; surgical emergency |
| Dissection Type B | Descending; medically managed; lower immediate mortality |
| Giant cell arteritis | >50 yrs; temporal headache + blindness; granulomatous; elevated ESR/IL-6; treat immediately with steroids |
| Takayasu | <50 yrs; Asian female; pulseless disease; renal HTN |
| PAN | Medium vessels; HBV-associated 30%; no pulmonary; no ANCA; beaded angiography; skip lesions |
| Kawasaki | Children; fever + rash + conjunctivitis + strawberry tongue; coronary aneurysms → IVIG + aspirin |
| GPA | c-ANCA (PR3); saddle nose + lungs + RPGN; rituximab/cyclophosphamide |
| EGPA | p-ANCA (MPO) 40%; asthma + eosinophilia + vasculitis |
| MPA | p-ANCA (MPO); no granulomas; RPGN + pulmonary hemorrhage |
| HSP (IgA vasculitis) | Children; IgA IC; purpura + arthritis + GI + renal |
| Primary Raynaud | Young females; intrinsic SMC hyperreactivity; benign |
| Secondary Raynaud | Underlying SLE/scleroderma/Buerger; progressive |
| Varicose veins | Valve incompetence; stasis; ulcers; phleboliths; esophageal varices from portal HTN |
| Trousseau syndrome | Migratory thrombophlebitis + occult cancer (pancreatic most classic) |
| SVC syndrome | Bronchogenic CA or lymphoma; facial/neck edema + dilated chest veins |
| Port-wine stain | Does NOT involute; grows with child; Sturge-Weber if V1 |
| Strawberry hemangioma | INVOLUTES by age 5-8; commonest childhood vascular tumor |
| Glomus tumor | Subungual; exquisitely painful |
| Bacillary angiomatosis | Bartonella; HIV patients; treat with erythromycin |
| Kaposi sarcoma | HHV-8; 4 forms; AIDS-KS most aggressive |
| Angiosarcoma | Stewart-Treves; vinyl chloride/Thorotrast/arsenic → hepatic; poor prognosis |
| Stent restenosis | Neointimal hyperplasia (SMC proliferation); DES reduce it but ↑ late thrombosis |
| Graft failure | Early: thrombosis; intermediate: intimal hyperplasia; late: atherosclerosis |
Source: Robbins & Cotran Pathologic Basis of Disease, 10th ed., Chapter 11 - The Blood Vessels