Now I have everything I need to write a comprehensive, textbook-grounded answer. Let me compile it.
Complete Guide to Pharmacology — From Foundations to Clinical Applications
Sources: Lippincott Illustrated Reviews Pharmacology, Katzung's Basic & Clinical Pharmacology 16th ed., Goodman & Gilman's Pharmacological Basis of Therapeutics
PART 1: WHAT IS PHARMACOLOGY?
Pharmacology is the science that studies how drugs interact with living systems — from molecules to entire organisms. It is divided into two inseparable pillars:
| Pillar | Question it Answers | Core Concern |
|---|
| Pharmacokinetics (PK) | What the body does to the drug | ADME |
| Pharmacodynamics (PD) | What the drug does to the body | Receptors, dose-response |
PART 2: PHARMACOKINETICS — ADME
A. Absorption
The process by which a drug enters the bloodstream. Governed by:
- Route of administration — IV bypasses absorption entirely (100% bioavailability); oral has first-pass metabolism
- pH and ionization — Weak acids absorb best in the stomach (low pH = nonionized form); weak bases absorb best in the small intestine (higher pH). Only the nonionized form crosses lipid membranes.
- Bioavailability (F) — Fraction of the administered dose that reaches systemic circulation unchanged
Example: A weakly basic drug (pKa 7.8) is mainly in the nonionized form at jejunal pH ~8.0, so it is best absorbed there. (Lippincott, p. 78)
B. Distribution
Once in blood, drugs spread into tissues depending on:
- Protein binding — Albumin binds acidic drugs; α1-acid glycoprotein binds basic drugs. Bound drug is pharmacologically inactive.
- Volume of Distribution (Vd) — How widely a drug spreads. High Vd = drug distributes extensively into tissues (chloroquine); Low Vd = drug stays in plasma (warfarin, heparin).
- Blood-Brain Barrier — Only lipid-soluble, non-protein-bound, small molecules penetrate the CNS.
C. Metabolism (Biotransformation)
Mainly hepatic. Two phases:
- Phase I — Oxidation, reduction, hydrolysis via CYP450 enzymes. Converts drug to a more polar (water-soluble) compound; may activate prodrugs (codeine → morphine) or deactivate drugs.
- Phase II — Conjugation with glucuronic acid, sulfate, acetate, amino acids → highly water-soluble conjugates → excreted in urine/bile.
First-pass effect: Orally absorbed drugs pass through the liver before reaching systemic circulation. Drugs with high first-pass (lidocaine, morphine) have very low oral bioavailability.
D. Excretion
- Renal — Glomerular filtration, tubular secretion, tubular reabsorption (pH-dependent). Alkalinizing urine traps acidic drugs (aspirin overdose treated with NaHCO₃).
- Biliary/fecal — Large molecular weight drugs (>300 Da) excreted via bile; can undergo enterohepatic recirculation.
Key PK Parameters
| Parameter | Meaning | Formula |
|---|
| Half-life (t½) | Time for plasma concentration to fall 50% | 0.693 × Vd / Cl |
| Clearance (Cl) | Volume of plasma cleared per unit time | Dose / AUC |
| Steady state | Reached after ~5 half-lives | - |
| Loading dose | Achieves therapeutic levels quickly | Vd × Cp (target) |
PART 3: RECEPTORS — THE COMPLETE PICTURE
What Is a Receptor?
A receptor is any biological macromolecule to which a drug binds to produce a measurable response. Most receptors are membrane-bound proteins that transduce extracellular signals into intracellular responses. Enzymes, nucleic acids, and structural proteins can also act as drug receptors.
"Pharmacodynamics describes the actions of a drug on the body. Most drugs exert effects, both beneficial and harmful, by interacting with specialized target macromolecules called receptors." (Lippincott, p. 82)
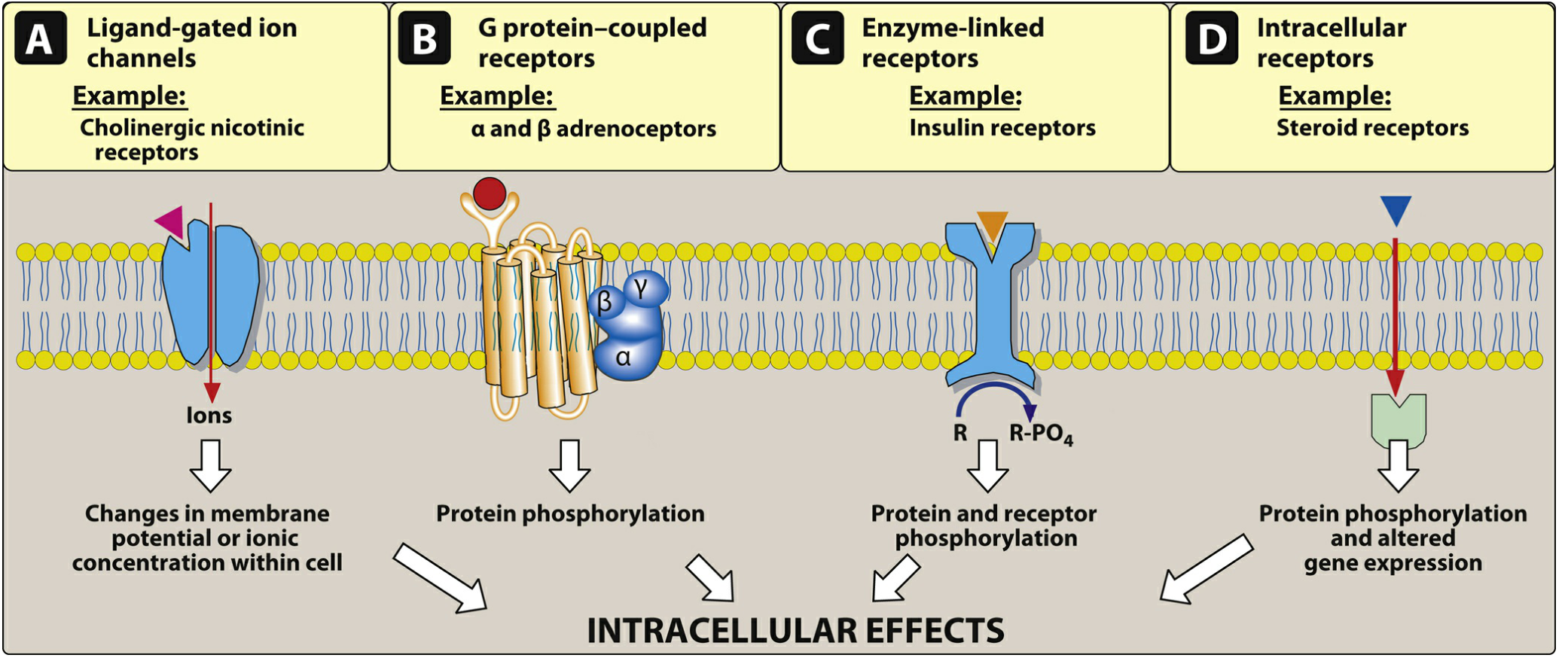
The Four Receptor Superfamilies
1. Ligand-Gated Ion Channels (Ionotropic Receptors) — Type I
- Structure: Transmembrane protein with an ion pore
- Speed: Milliseconds — fastest signaling
- Mechanism: Drug binds → channel opens → ions flow
- Examples:
- Nicotinic ACh receptor (nAChR): Na⁺ influx / K⁺ efflux → action potential in muscle/neurons
- GABA-A receptor: Cl⁻ influx → hyperpolarization → inhibition (benzodiazepines and barbiturates act here)
- Glutamate (NMDA) receptor: Na⁺/Ca²⁺ influx → excitation (ketamine blocks this)
- Clinical drugs: Benzodiazepines (enhance GABA-A), succinylcholine (nAChR agonist), local anesthetics (block voltage-gated Na⁺ channels)
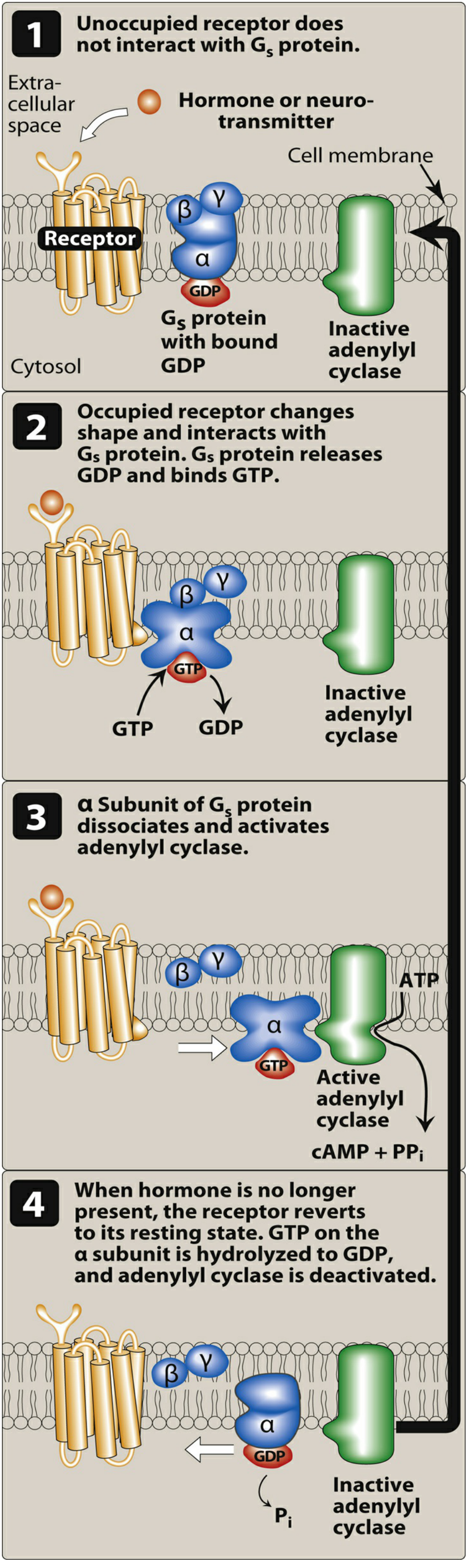
2. G Protein-Coupled Receptors (GPCRs) — Type II
The largest and most drug-targeted receptor family (~30–35% of all drug targets).
- Structure: 7 transmembrane helices; intracellular G protein (α, β, γ subunits)
- Speed: Seconds to minutes
G Protein Types and their second messengers:
| G Protein | Effect on Adenylyl Cyclase | Second Messenger | Examples |
|---|
| Gs | ↑ (activates) | ↑ cAMP → PKA | β1-adrenoceptor (heart), β2 (bronchi) |
| Gi | ↓ (inhibits) | ↓ cAMP | α2-adrenoceptors, muscarinic M2 |
| Gq | → Phospholipase C | ↑ IP3 + DAG → PKC | α1-adrenoceptors, muscarinic M1/M3, H1 |
Why this matters clinically: A drug that activates Gs in the heart (e.g., epinephrine on β1) increases heart rate and contractility. That same epinephrine on β2 in the lung (also Gs) causes bronchodilation. The same signal cascade, different tissue = different clinical effect.
GPCR Examples by clinical area:
- Adrenoceptors (α1, α2, β1, β2, β3) — epinephrine, norepinephrine, salbutamol
- Muscarinic receptors (M1–M5) — acetylcholine, atropine
- Histamine receptors (H1, H2, H3, H4) — histamine, cetirizine, ranitidine
- Dopamine receptors (D1–D5) — levodopa, haloperidol, clozapine
- Opioid receptors (μ, κ, δ) — morphine, naloxone
- Serotonin receptors (5-HT1–7) — SSRIs, triptans, ondansetron
3. Enzyme-Linked Receptors (Receptor Tyrosine Kinases) — Type III
- Structure: Single transmembrane domain; intracellular kinase domain
- Mechanism: Ligand binds → receptor dimerizes → autophosphorylation → downstream phosphorylation cascade
- Speed: Minutes to hours
- Examples:
- Insulin receptor — phosphorylates IRS proteins → GLUT4 translocation → glucose uptake
- Growth hormone receptor — JAK/STAT pathway
- EGF receptor — targeted by drugs like erlotinib (cancer)
- VEGF receptor — targeted by bevacizumab (antiangiogenic therapy)
4. Intracellular (Nuclear) Receptors — Type IV
- Structure: Cytoplasmic or nuclear proteins; act as transcription factors
- Ligand requirement: Must be lipid-soluble to cross the cell membrane
- Speed: Hours to days — gene expression changes
- Second messengers: None; direct DNA binding
- Examples:
- Glucocorticoid receptor — cortisol, prednisone, dexamethasone
- Mineralocorticoid receptor — aldosterone, fludrocortisone
- Thyroid hormone receptor — T3, T4
- Sex hormone receptors — estrogen (ER), androgen (AR), progesterone (PR)
- Retinoic acid receptor — Vitamin A, isotretinoin
Steroid hormones diffuse across membranes, bind cytoplasmic receptors → receptor-hormone complex translocates to nucleus → binds hormone response elements on DNA → alters gene transcription.
Receptor States — Active vs. Inactive
Receptors exist in equilibrium between two states: R (inactive) ⇌ *R (active)**.
| Drug Type | Binding | Shifts R→R* | Effect |
|---|
| Full agonist | Yes | Maximum | Full response (Emax) |
| Partial agonist | Yes | Partial | Submaximal response even at full dose |
| Antagonist | Yes | None | Blocks agonist; no effect alone |
| Inverse agonist | Yes | Reverses baseline R* | Decreases baseline activity below resting level |
Types of Antagonism
-
Competitive (reversible): Binds same site as agonist; shifts dose-response curve right (↑ EC50), Emax unchanged. Overcome by increasing agonist concentration. Example: atropine blocking ACh at muscarinic receptors
-
Irreversible (noncompetitive): Binds covalently → permanently reduces Emax; EC50 unchanged. Cannot be overcome. Example: phenoxybenzamine at α-receptors
-
Allosteric antagonism: Binds a different site; changes receptor conformation → blocks agonist effect. Also ↓ Emax. Example: picrotoxin inside the GABA-A chloride channel
-
Functional (physiological) antagonism: Two drugs act at separate receptors with opposing effects. Example: epinephrine (β2 → bronchodilation) opposes histamine (H1 → bronchoconstriction)
Dose-Response Relationships
- Graded dose-response: As dose increases, response increases — used to measure potency (EC50) and efficacy (Emax).
- EC50 — dose producing 50% of maximum response = measure of potency (lower EC50 = more potent)
- Emax — maximum possible response = measure of efficacy
- Therapeutic Index (TI) = TD50 / ED50. A wide TI (e.g., penicillin) means more safety margin; narrow TI (e.g., digoxin, lithium, warfarin) requires therapeutic drug monitoring.
PART 4: HOW RECEPTORS INTERCONNECT EVERY CHAPTER OF PHARMACOLOGY
This is the KEY insight: every pharmacology chapter is just a different receptor or enzyme being targeted. Here's the map:
| Chapter/System | Receptor / Target | Drugs |
|---|
| Autonomic/ANS | α1, α2, β1, β2, M1-M5, nAChR | Epinephrine, atropine, propranolol, prazosin |
| Cardiovascular | β1, AT1, L-type Ca²⁺, Na⁺/K⁺-ATPase | Metoprolol, losartan, amlodipine, digoxin |
| Pulmonary | β2, M3, LTR, H1 | Salbutamol, ipratropium, montelukast |
| CNS/Psychiatry | D2, 5-HT2A, GABA-A, NMDA, μ-opioid | Haloperidol, diazepam, ketamine, morphine |
| Endocrine | Insulin receptor, thyroid R, glucocorticoid R | Insulin, levothyroxine, dexamethasone |
| GI | H2, M3, 5-HT4, PPI (H⁺/K⁺-ATPase) | Ranitidine, hyoscine, metoclopramide, omeprazole |
| Inflammation | COX-1/2 (enzyme), glucocorticoid R | Aspirin/NSAIDs, prednisone |
| Oncology | RTKs (EGFR, VEGFR), ER, AR | Erlotinib, tamoxifen, enzalutamide |
| Infection | Bacterial ribosomes, DNA gyrase, cell wall | Antibiotics (not receptor-based but same concept) |
PART 5: DRUG GENERATIONS — WHAT DOES 1st, 2nd, 3rd GENERATION MEAN?
"Generation" refers to when a drug class was developed and what improvements were made. Each successive generation aims for greater selectivity, fewer side effects, or better pharmacokinetics.
EXAMPLE 1: ANTIHISTAMINES (H1 Blockers)
All generations block the H1 receptor (a Gq-coupled GPCR). The difference is selectivity and CNS penetration.
| Generation | Examples | CNS Penetration | Sedation | Other Effects |
|---|
| 1st (1940s–60s) | Diphenhydramine (Benadryl), Chlorpheniramine, Promethazine, Hydroxyzine | High — cross BBB | Strong sedation | Anticholinergic (dry mouth, urinary retention, blurred vision), anti-motion sickness |
| 2nd (1980s–90s) | Loratadine, Cetirizine, Fexofenadine | Low — P-glycoprotein efflux at BBB | Minimal sedation | Less anticholinergic; preferred for allergy |
| 3rd (metabolites of 2nd) | Desloratadine (from loratadine), Levocetirizine (from cetirizine) | Very low | Essentially none | More potent, longer-acting, more selective |
"The second-generation H1 antagonists are used mainly for the treatment of allergic rhinitis and chronic urticaria... sedation occurred in only about 7% of subjects taking the second-generation agents [vs ~50% with first-generation]." (Katzung, p. 441)
Why does the 1st generation cause sedation? Because it penetrates the CNS and blocks central H1 receptors involved in wakefulness. The 2nd/3rd generations are excluded from the CNS by the P-glycoprotein pump and are more polar.
Why does 1st generation cause dry mouth/urinary retention? Because 1st-generation antihistamines also block muscarinic M receptors (anticholinergic activity) — they are non-selective. 2nd generation has negligible anticholinergic action.
EXAMPLE 2: BETA-BLOCKERS (β-Adrenoceptor Antagonists)
All block β-adrenoceptors (Gs-coupled GPCRs). Generations differ in selectivity and additional properties.
| Generation | Examples | β1 Selectivity | Extra Features | Side Effect Profile |
|---|
| 1st | Propranolol, Timolol, Nadolol | Non-selective (β1 + β2) | None | Bronchoconstriction (β2 block), mask hypoglycemia, peripheral vasoconstriction |
| 2nd | Metoprolol, Atenolol, Bisoprolol | Cardioselective (β1 > β2) | None | Less bronchoconstriction; safer in COPD/asthma (relatively) |
| 3rd | Carvedilol, Labetalol, Nebivolol | Variable | + α1 blockade (vasodilation) or + NO release (nebivolol) | Additional hypotension from α1 block; nebivolol has least metabolic side effects |
"The third-generation beta blocker drugs are cardioselective and have additional vasodilatory properties (nebivolol), used alone or in combination with α-receptor blockade (carvedilol)." (Harrison's, block 30)
"Cardioselective β-blockers that only block β1 receptors, such as metoprolol and atenolol, are preferred. All β-blockers are nonselective at high doses and can inhibit β2 receptors." (Lippincott, p. 418)
Clinical implication: A 1st generation drug like propranolol blocks β2 in the lungs → bronchoconstriction — dangerous in asthma. A 2nd generation like metoprolol at usual doses spares β2 → safer for the lungs. But it still blocks β1 in the heart → slows HR and reduces contractility.
EXAMPLE 3: ANTIPSYCHOTICS (Dopamine/Serotonin Antagonists)
| Generation | Examples | Receptor Target | Key Issue |
|---|
| 1st (FGA / Typical) | Haloperidol, Chlorpromazine, Fluphenazine | Dopamine D2 block | Extrapyramidal symptoms (EPS): parkinsonism, dystonia, tardive dyskinesia, akathisia |
| 2nd (SGA / Atypical) | Risperidone, Olanzapine, Quetiapine, Aripiprazole, Clozapine | D2 + 5-HT2A block | Less EPS, but metabolic syndrome (weight gain, diabetes, dyslipidemia) |
| 3rd (partial agonists) | Aripiprazole, Brexpiprazole, Cariprazine | Partial D2/D3 agonist + 5-HT1A partial agonist + 5-HT2A antagonist | Least metabolic effects; no complete dopamine blockade |
"First-generation antipsychotics are more likely to be associated with movement disorders known as extrapyramidal symptoms (EPS)... The second-generation antipsychotic drugs (SGAs) have a lower incidence of EPS but are associated with a higher risk of metabolic adverse effects, such as diabetes, hypercholesterolemia, and weight gain. The second-generation drugs owe their unique activity to blockade of both serotonin 5-HT2A and dopamine D2 receptors." (Lippincott, p. 612–613)
Why does 1st generation cause EPS? D2 receptor blockade in the nigrostriatal dopamine pathway → motor side effects. The 2nd generation's additional 5-HT2A blockade reduces dopamine release in the striatum from a different angle, reducing EPS.
EXAMPLE 4: CEPHALOSPORINS (Antibiotic Generations)
Here "generation" indicates spectrum of antimicrobial activity, not receptor-based:
| Generation | Examples | Spectrum | Key Use |
|---|
| 1st | Cefazolin, Cefalexin | Gram-positive dominant | Skin/soft tissue, surgical prophylaxis |
| 2nd | Cefuroxime, Cefaclor | Extended Gram-negative coverage | RTI, sinusitis |
| 3rd | Ceftriaxone, Cefotaxime, Ceftazidime | Broad Gram-negative; ↓ Gram-positive | Meningitis, sepsis, Pseudomonas (ceftazidime) |
| 4th | Cefepime | Broad spectrum including Pseudomonas | Nosocomial infections |
| 5th | Ceftaroline | + MRSA activity | MRSA pneumonia/skin infections |
PART 6: SAME DRUG — DIFFERENT RECEPTOR IN DIFFERENT SYSTEM = DIFFERENT USE AND SIDE EFFECT
This is one of the most powerful concepts in pharmacology. The same drug hits multiple receptors or the same receptor in different organs — each with distinct outcomes.
EPINEPHRINE — One Drug, Many Receptors, Many Uses
| Receptor Activated | Location | Effect | Clinical Use |
|---|
| β1 (Gs → ↑ cAMP) | Heart | ↑ HR, ↑ contractility | Cardiac arrest (IV) |
| β2 (Gs → ↑ cAMP) | Bronchial smooth muscle | Bronchodilation | Anaphylaxis (SC/IM) |
| α1 (Gq → IP3/DAG) | Vascular smooth muscle | Vasoconstriction | Shock (IV), prolongs local anesthesia |
| α2 (Gi → ↓ cAMP) | Presynaptic nerve terminals | ↓ Norepinephrine release | Negative feedback |
Side effects: Hypertension (α1), tachycardia/arrhythmia (β1), tremor (β2), anxiety (CNS)
PROPRANOLOL — Non-selective β-blocker
| Receptor Blocked | System | Effect | Clinical Use |
|---|
| β1 | Heart | ↓ HR, ↓ contractility, ↓ AV conduction | Hypertension, angina, arrhythmia |
| β1 | Kidney (JG cells) | ↓ Renin release → ↓ BP | Hypertension |
| β2 | Bronchi | Bronchoconstriction | Side effect — contraindicated in asthma |
| β2 | Liver | Masks hypoglycemia (blocks glycogenolysis) | Side effect in diabetes |
| β (CNS) | Brain | Reduces anxiety/tremor | Anxiety, essential tremor, performance anxiety |
| β (Eye) | Ciliary body | ↓ Aqueous humor production | Glaucoma (timolol eye drops) |
ATROPINE — Muscarinic Antagonist (same receptor M everywhere, different organ effects)
| Receptor Blocked | Organ | Normal muscarinic effect | Atropine Effect | Use |
|---|
| M2 | Heart | Slows HR (vagal) | ↑ HR (tachycardia) | Bradycardia, organophosphate poisoning |
| M3 | Smooth muscle (GI, bladder) | Contraction | Relaxation → ileus, urinary retention | IBS, bladder spasm |
| M3 | Glands (salivary, sweat) | Secretion | ↓ Secretion → dry mouth, anhidrosis | Pre-operative drying agent |
| M3 | Bronchi | Bronchoconstriction | Bronchodilation | COPD (ipratropium = selective M3) |
| M3 | Eye (pupillary sphincter) | Miosis (pupil constriction) | Mydriasis (dilation) | Eye exam, uveitis |
| M3 | Eye (ciliary muscle) | Accommodation (near vision) | Cycloplegia (blurred near vision) | Side effect |
Mnemonic for atropine toxicity: "Hot as a hare, dry as a bone, red as a beet, blind as a bat, mad as a hatter" — hyperthermia, anhidrosis, flushing, mydriasis, confusion.
MORPHINE — μ-Opioid Receptor Agonist
| Location of μ Receptor | Effect of Morphine | Use or Side Effect |
|---|
| CNS (PAG, thalamus) | Analgesia | Pain relief |
| Limbic system | Euphoria | Addiction potential |
| Medullary respiratory center | ↓ Respiratory drive | Respiratory depression (main cause of death in overdose) |
| Cough center | Suppresses cough | Antitussive |
| GI (myenteric plexus) | ↓ Peristalsis | Constipation, used for diarrhea (loperamide = peripheral opioid) |
| Bladder detrusor | ↓ Bladder tone | Urinary retention |
| Mast cells | Histamine release | Itching, hypotension |
| Eye | Miosis | Pin-point pupils (diagnostic in overdose) |
METFORMIN — Non-receptor, but illustrates system-specific action
Acts via AMPK activation (AMP-activated protein kinase) → inhibits hepatic gluconeogenesis primarily:
- Liver: ↓ gluconeogenesis (main effect) → lowers fasting glucose
- GI: Delayed absorption → weight-neutral, GI side effects (nausea, diarrhea)
- No β-cell stimulation → no hypoglycemia
- Kidney: Lactic acidosis risk if GFR <30 (reduced clearance)
PART 7: RECEPTOR INTERCONNECTIONS — THE BIG PICTURE
Understanding the ANS (Autonomic Nervous System) receptor map ties everything together:
SYMPATHETIC ("fight or flight")
└─ Norepinephrine / Epinephrine
├─ α1 (Gq) → vascular constriction, mydriasis, urinary retention
├─ α2 (Gi) → presynaptic inhibition, ↓ insulin, ↓ NE release
├─ β1 (Gs) → ↑ HR, ↑ contractility, ↑ renin, lipolysis
├─ β2 (Gs) → bronchodilation, vasodilation, glycogenolysis, uterine relaxation
└─ β3 (Gs) → lipolysis in adipose
PARASYMPATHETIC ("rest and digest")
└─ Acetylcholine
├─ M1 (Gq) → CNS cognition, gastric acid (ECL cells)
├─ M2 (Gi) → ↓ HR, ↓ conduction (SA/AV nodes)
├─ M3 (Gq) → gland secretion, smooth muscle contraction, bronchoconstriction, miosis
├─ M4 (Gi) → CNS modulation
└─ M5 (Gq) → CNS/dopamine modulation
SOMATIC / NEUROMUSCULAR
└─ ACh on Nicotinic (nAChR) — ion channel (Na⁺/K⁺) → skeletal muscle contraction
Every drug class in autonomic pharmacology is just targeting one node of this map:
- Phenylephrine (α1 agonist) → nasal decongestant, raises BP
- Clonidine (α2 agonist) → lowers BP (reduces central NE release)
- Salbutamol (β2 agonist) → asthma bronchodilator
- Atenolol (β1 blocker) → lowers HR/BP
- Pilocarpine (M agonist) → treats glaucoma (increases aqueous drainage)
- Atropine (M antagonist) → treats bradycardia, dilates pupils
- Neostigmine (AChE inhibitor) → raises ACh at all muscarinic and nicotinic sites → treats myasthenia gravis, reverses neuromuscular blockade
PART 8: THERAPEUTIC INDEX AND DRUG SAFETY
| Drug | Therapeutic Index | Clinical Need |
|---|
| Penicillin | Very wide | No monitoring needed |
| Digoxin | Narrow | Serum level monitoring |
| Warfarin | Narrow | INR monitoring |
| Lithium | Narrow | Serum lithium monitoring |
| Phenytoin | Narrow | Serum level, nonlinear kinetics |
| Aminoglycosides | Narrow | Peak and trough levels |
| Theophylline | Narrow | Serum levels; metabolized by CYP1A2 |
SUMMARY: HOW TO THINK ABOUT ANY DRUG
When you encounter any drug, ask these 5 questions:
- What receptor/enzyme does it target? → determines mechanism
- Is it an agonist, antagonist, partial agonist? → determines direction of effect
- What is the receptor type (GPCR, ion channel, kinase, nuclear)? → determines speed and nature of signal
- Where is that receptor expressed? → predicts both therapeutic effects AND side effects
- What are its PK properties (lipid soluble? first pass? half-life? renal/hepatic clearance)? → determines dosing, routes, drug interactions
Every chapter of pharmacology — from cardiovascular to psychiatry to endocrinology to oncology — follows this exact same framework. The generations (1st, 2nd, 3rd) represent the evolution toward more selective, safer, and longer-acting drugs that hit the right receptor in the right organ without the off-target effects that caused problems in earlier molecules.
Sources: Lippincott Illustrated Reviews: Pharmacology | Katzung's Basic and Clinical Pharmacology 16th Ed. | Goodman & Gilman's Pharmacological Basis of Therapeutics | Harrison's Principles of Internal Medicine 22nd Ed.