Cardiac biomarkers values and significance in ACS and other disease causing elevated values as per harrison
Cardiac Biomarkers in ACS — Harrison's Principles of Internal Medicine, 22E (2025)
1. Fundamental Principle
A rise and/or fall in cardiac biomarker values with at least one value above the 99th percentile upper reference limit.
2. Classification of Elevated cTn (Figure 15-3)
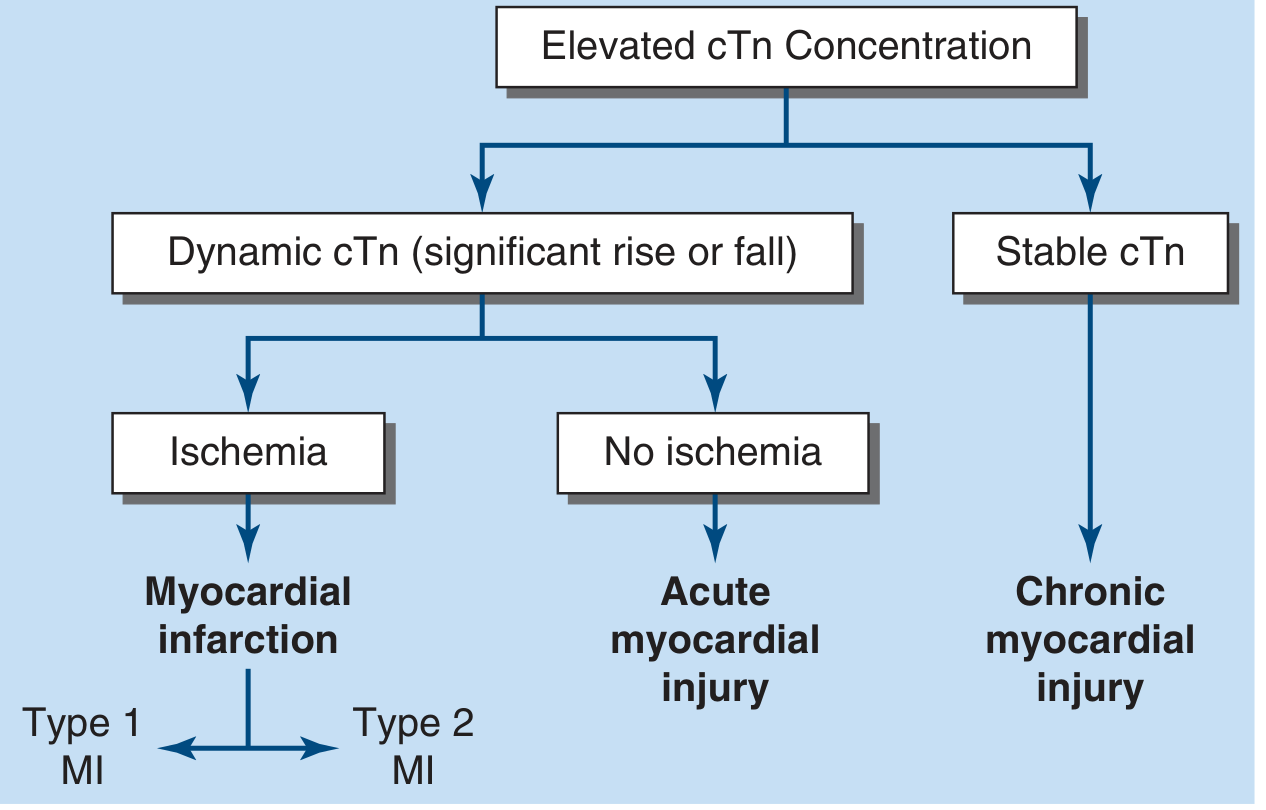
| Pattern | Interpretation |
|---|---|
| Dynamic cTn (significant rise or fall) + Ischemia | Myocardial Infarction (Type 1 or Type 2 MI) |
| Dynamic cTn + No ischemia | Acute myocardial injury (non-ischemic) |
| Stable cTn | Chronic myocardial injury |
3. Biomarkers — Individual Details
A. Cardiac Troponin I (cTnI) and Troponin T (cTnT) ⭐ Preferred
- Structure: cTnT and cTnI have amino-acid sequences that differ from skeletal muscle isoforms, enabling highly specific monoclonal antibody–based assays.
- Measurement: High-sensitivity (hs-cTn) assays are preferred over conventional assays for all suspected ACS.
- Cutoff: >99th percentile of reference population = abnormal.
- Rise in STEMI: cTn rises to 20–50× the upper reference limit in "classic" large MI.
- Temporal profile (STEMI):
- Detectable: ~2–4 h after onset
- Peak: ~12–24 h (NSTEMI); earlier peak if reperfusion achieved (washout effect)
- Elevated for: at least 7–10 days after STEMI
- NSTEMI: Characteristic temporal rise peaking at 12–24 h after symptom onset, then gradually decreasing; direct relationship between degree of elevation and mortality.
- High-sensitivity assays allow:
- Rapid rule-out protocols using serial testing over 1–2 h
- In patients >2–3 h after symptom onset: very low hs-cTn at presentation alone may exclude MI (NPV >99%)
- The 1-h rapid rule-out algorithm (no abnormal elevation of hsTn at 0 or 1 h) recommended by recent practice guidelines
- Measurement timing: At presentation, then repeat at 1–3 h (hs-cTn) or 3–6 h (conventional cTn); further measurements if clinical uncertainty persists.
B. Creatine Kinase-MB (CK-MB)
- Now secondary — not cost-effective to measure alongside cardiac-specific troponin.
- Relative index: CK-MB mass / CK activity ≥2.5 suggests (but is not diagnostic of) a myocardial source.
- Key remaining niche: Because CK-MB declines more rapidly than cTn after AMI onset, it remains useful for detecting early reinfarction during the period when cTn remains elevated from the index event.
- Microinfarction: cTn elevations occur above the upper reference limit in microinfarction even when CK-MB is still within the normal range — an important diagnostic advantage of hs-cTn.
C. Myoglobin
- Released earliest (cytoplasmic pool; small molecular weight).
- Blood levels rise quickly above the cutoff — earliest marker.
- No cardiac specificity — rendered obsolete by troponin assays.
Biomarker Kinetics (Figure 286-4)
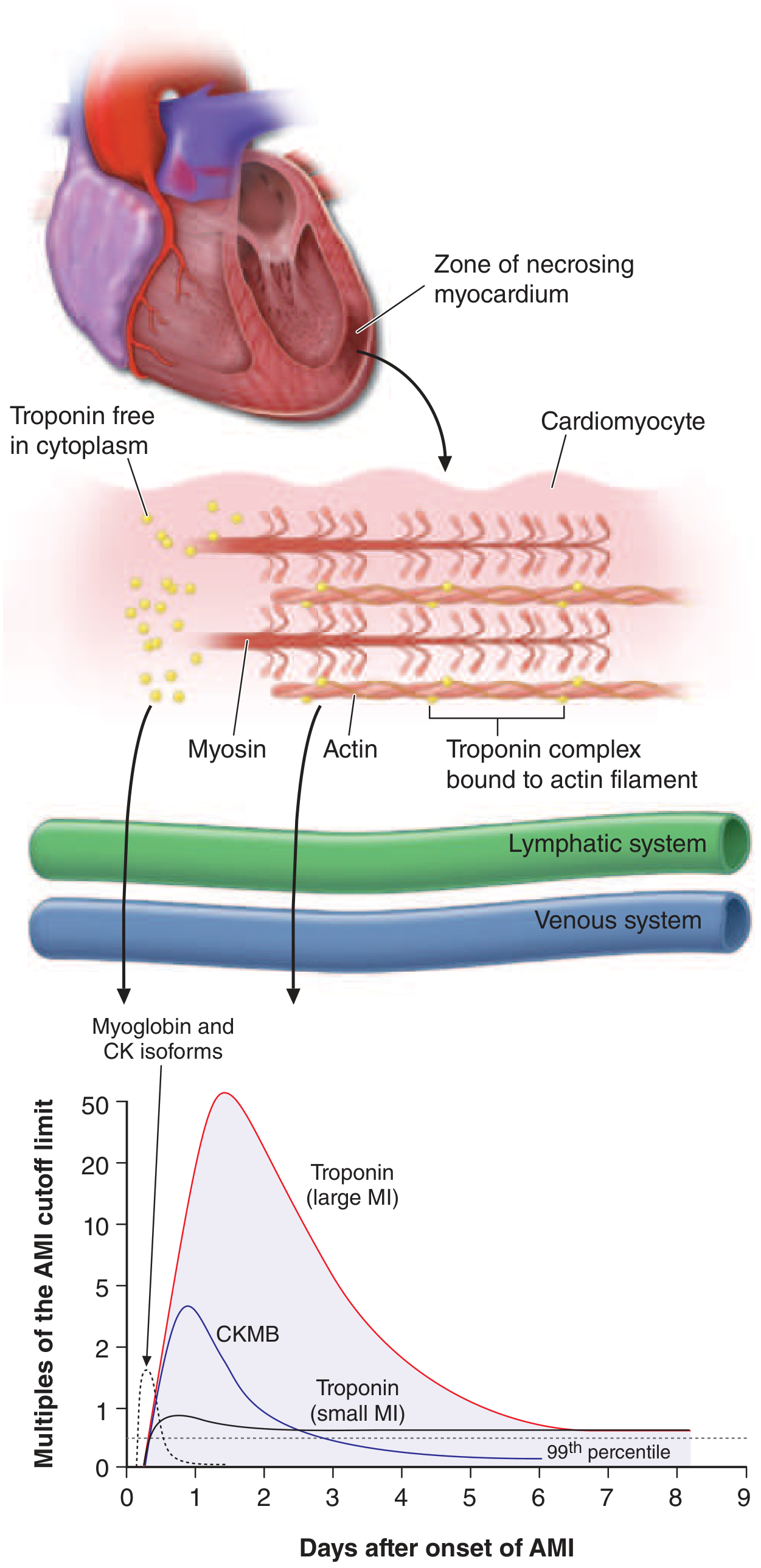
| Biomarker | Rises above cutoff | Peak | Returns to normal |
|---|---|---|---|
| Myoglobin / CK isoforms | ~1–4 h | ~6–9 h | ~24 h |
| CK-MB | ~4–6 h | ~18–24 h | ~48–72 h |
| Troponin (small MI) | ~4–6 h | ~12–24 h | ~5–7 days |
| Troponin (large MI) | ~4–6 h | ~24–48 h | up to 7–10+ days |
4. Other Laboratory Markers
- D-dimer: Useful to exclude pulmonary embolism in the differential of acute chest pain.
- BNP / NT-proBNP: Useful when considered alongside clinical history and examination for the diagnosis of heart failure; multimarker strategies including BNP alongside hsTn are gaining favor for risk stratification in NSTE-ACS.
- WBC: Polymorphonuclear leukocytosis appears within hours, persists 3–7 days, WBC up to 12,000–15,000/μL — nonspecific reaction to myocardial injury.
- ESR: Rises more slowly than WBC, peaks during the first week, may remain elevated 1–2 weeks.
5. Causes of Elevated Cardiac Troponin — Beyond ACS
Myocardial Injury Related to Acute MI
- Atherosclerotic plaque disruption or erosion with thrombosis
Myocardial Ischemia (O₂ Supply/Demand Imbalance — Type 2 MI territory)
| Category | Examples |
|---|---|
| Reduced perfusion | Coronary artery spasm, microvascular dysfunction, coronary embolism, coronary artery dissection |
| Bradyarrhythmia | Sustained bradyarrhythmia |
| Hemodynamic | Hypotension or shock |
| Respiratory | Respiratory failure |
| Hematologic | Severe anemia |
| Increased demand | Sustained tachyarrhythmia, severe hypertension |
Other Cardiac Causes
- Heart failure
- Myocarditis
- Cardiomyopathy (any type)
- Takotsubo syndrome (stress cardiomyopathy)
- Recent coronary revascularization
- Cardiac procedures other than revascularization
- Catheter ablation
- Defibrillator shocks
- Cardiac contusion
Systemic (Non-Cardiac) Causes
- Sepsis
- Chronic kidney disease (reduced clearance + chronic myocardial injury)
- Stroke / subarachnoid hemorrhage (neurogenic myocardial injury)
- Pulmonary embolism
- Infiltrative diseases (e.g., amyloidosis, sarcoidosis)
- Chemotherapeutic agents (cardiotoxicity)
- Critical illness
- Strenuous exercise
Source: Reproduced with permission from K Thygesen et al: Fourth Universal Definition of Myocardial Infarction (2018). Circulation 138:2231, 2018.
6. Critical Clinical Distinctions
| Feature | MI Diagnosis | Non-Ischemic Injury |
|---|---|---|
| Pattern | Dynamic (rise and/or fall) | Dynamic but no ischemia (myocarditis, Takotsubo) |
| Cause | Must be ischemic | Other non-ischemic insults |
| Chronic elevation | Stable cTn — NOT MI | CKD, structural heart disease, interfering antibodies |
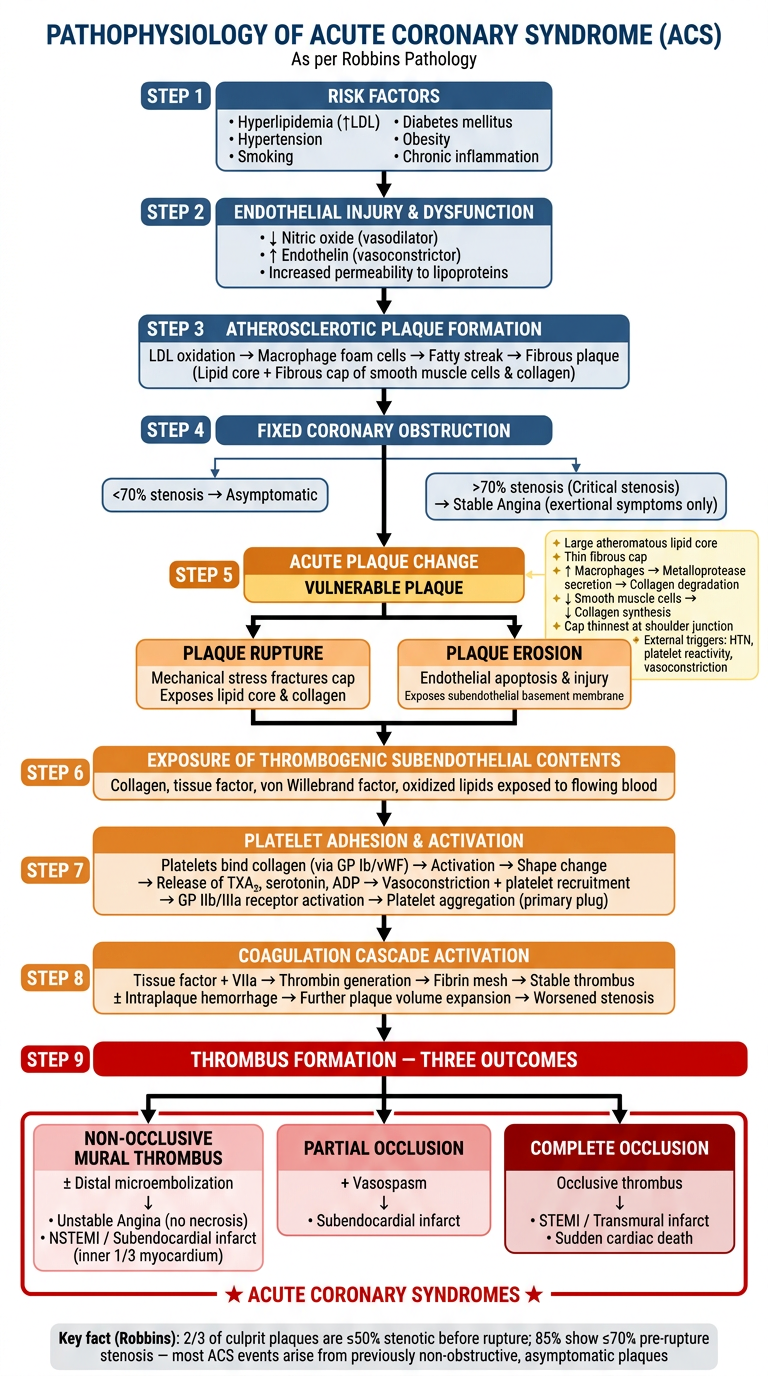
Pathophysiology of Acs in seqence in diagram or flowchart form as per robbins
A detailed medical pathophysiology flowchart titled "Pathophysiology of Acute Coronary Syndrome (ACS) — Robbins" showing the following sequential steps in a vertical flow with connecting arrows and branching outcomes: TOP SECTION - "ATHEROSCLEROSIS & PLAQUE FORMATION": Box 1: "Risk Factors: Hyperlipidemia, HTN, Smoking, Diabetes, Inflammation" ↓ arrow Box 2: "Endothelial Injury & Dysfunction → LDL oxidation → Foam cell formation → Fatty streak → Fibrous plaque (atheroma)" ↓ arrow Box 3: "FIXED CORONARY OBSTRUCTION" with two branches: - Left branch: "<70% stenosis → Asymptomatic" - Right branch: ">70% stenosis (Critical stenosis) → Stable Angina (symptoms only on exertion)" ↓ arrow continuing center Box 4: "VULNERABLE PLAQUE" with feature list in a side box (light yellow): • Large atheromatous lipid core • Thin fibrous cap • ↑ Macrophage inflammation • ↓ Smooth muscle cells • Metalloprotease-mediated collagen degradation • Cap thinnest at shoulder (junction with normal wall) MIDDLE SECTION - "ACUTE PLAQUE CHANGE" (highlighted in orange/red): Two pathways side by side connected to Vulnerable Plaque: Left path: "PLAQUE RUPTURE" (mechanical stress, HTN, vasoconstriction) Right path: "PLAQUE EROSION" (endothelial apoptosis, inflammation, toxic exposure) Both merge to: ↓ arrow Box 5: "Exposure of thrombogenic subendothelial collagen & lipid core" ↓ arrow Box 6: "Platelet Adhesion & Activation → Platelet aggregation → Vasoactive mediators (TXA₂, serotonin) → Vasoconstriction" ↓ arrow Box 7: "Coagulation Cascade Activation → Thrombin generation → Fibrin clot formation" LOWER SECTION - "THROMBUS OUTCOMES" (three outcome branches): Branch 1 (Left): "Mural (Nonocclusive) Thrombus ± Distal Embolization" → Unstable Angina (no myocyte necrosis) → NSTEMI / Subendocardial Infarct Branch 2 (Center): "Partially Occlusive Thrombus + Vasospasm" → Subendocardial Infarct (innermost myocardium) Branch 3 (Right): "Completely Occlusive Thrombus" → STEMI / Transmural Infarct → Sudden Cardiac Death Bottom box spanning all branches labeled "ACUTE CORONARY SYNDROMES" in bold red. Small note box at bottom: "Note: 2/3 of culprit lesions are ≤50% stenotic before rupture; 85% are ≤70% — most are asymptomatic prior to ACS event" Use a clean white background, blue/dark navy boxes for main steps, light orange highlight for the acute plaque change section, red for the ACS outcomes box, connecting arrows in dark grey, clear readable font. Professional medical textbook style.
Pathophysiology of Acs in seqence in diagram or flowchart form as per robbins
A clean, detailed, professional medical pathophysiology sequential flowchart titled "PATHOPHYSIOLOGY OF ACUTE CORONARY SYNDROME (ACS)" with subtitle "As per Robbins Pathology" on a white background. The flowchart flows top-to-bottom with labeled numbered steps and color-coded sections. Use dark blue rounded rectangles for main steps, light yellow side-annotation boxes, orange for the "acute plaque change" section, and red for the final ACS outcomes box. Use bold black arrows between steps. STEP 1 (dark blue box, top center): "RISK FACTORS" • Hyperlipidemia (↑LDL) • Hypertension • Smoking • Diabetes mellitus • Obesity • Chronic inflammation ↓ arrow STEP 2 (dark blue box): "ENDOTHELIAL INJURY & DYSFUNCTION" • ↓ Nitric oxide (vasodilator) • ↑ Endothelin (vasoconstrictor) • Increased permeability to lipoproteins ↓ arrow STEP 3 (dark blue box): "ATHEROSCLEROTIC PLAQUE FORMATION" LDL oxidation → Macrophage foam cells → Fatty streak → Fibrous plaque (Lipid core + Fibrous cap of smooth muscle cells & collagen) ↓ arrow STEP 4 (dark blue box): "FIXED CORONARY OBSTRUCTION" [Two side branches here:] LEFT BRANCH: "<70% stenosis → Asymptomatic" RIGHT BRANCH: ">70% stenosis (Critical stenosis) → Stable Angina (exertional symptoms only)" [Center arrow continues down] ↓ arrow STEP 5 (orange/amber box — highlighted section titled "ACUTE PLAQUE CHANGE"): "VULNERABLE PLAQUE" [Yellow annotation box to the right listing vulnerability features:] ✦ Large atheromatous lipid core ✦ Thin fibrous cap ✦ ↑ Macrophages → Metalloprotease secretion → Collagen degradation ✦ ↓ Smooth muscle cells → ↓ Collagen synthesis ✦ Cap thinnest at shoulder junction ✦ External triggers: HTN, platelet reactivity, vasoconstriction [Two parallel arrows from STEP 5 side by side:] LEFT PATH: RIGHT PATH: "PLAQUE RUPTURE" "PLAQUE EROSION" Mechanical stress fractures cap Endothelial apoptosis & injury Exposes lipid core & collagen Exposes subendothelial basement membrane [Both merge into single arrow below] ↓ arrow STEP 6 (orange box): "EXPOSURE OF THROMBOGENIC SUBENDOTHELIAL CONTENTS" Collagen, tissue factor, von Willebrand factor, oxidized lipids exposed to flowing blood ↓ arrow STEP 7 (orange box): "PLATELET ADHESION & ACTIVATION" Platelets bind collagen (via GP Ib/vWF) → Activation → Shape change → Release of TXA₂, serotonin, ADP → Vasoconstriction + platelet recruitment → GP IIb/IIIa receptor activation → Platelet aggregation (primary plug) ↓ arrow STEP 8 (orange box): "COAGULATION CASCADE ACTIVATION" Tissue factor + VIIa → Thrombin generation → Fibrin mesh → Stable thrombus ± Intraplaque hemorrhage → Further plaque volume expansion → Worsened stenosis ↓ arrow STEP 9 (red box, bold text): "THROMBUS FORMATION — THREE OUTCOMES" [Three branches spread out:] LEFT BOX (pink): "NON-OCCLUSIVE MURAL THROMBUS ± Distal microembolization ↓ • Unstable Angina (no necrosis) • NSTEMI / Subendocardial infarct (inner 1/3 myocardium)" CENTER BOX (light red): "PARTIAL OCCLUSION + Vasospasm ↓ • Subendocardial infarct" RIGHT BOX (dark red, white text): "COMPLETE OCCLUSION Occlusive thrombus ↓ • STEMI / Transmural infarct • Sudden cardiac death" [All three boxes enclosed in a bold red border labeled:] "★ ACUTE CORONARY SYNDROMES ★" BOTTOM NOTE BOX (light grey): "Key fact (Robbins): 2/3 of culprit plaques are ≤50% stenotic before rupture; 85% show ≤70% pre-rupture stenosis — most ACS events arise from previously non-obstructive, asymptomatic plaques" The overall layout is tall portrait orientation, very readable, with clear step numbers on each box. Medical textbook diagram style.
Pathophysiology of ACS — Robbins Pathology
Custom Sequential Flowchart
Original Robbins Figure 12.9 (Robbins & Cotran, Pathologic Basis of Disease)
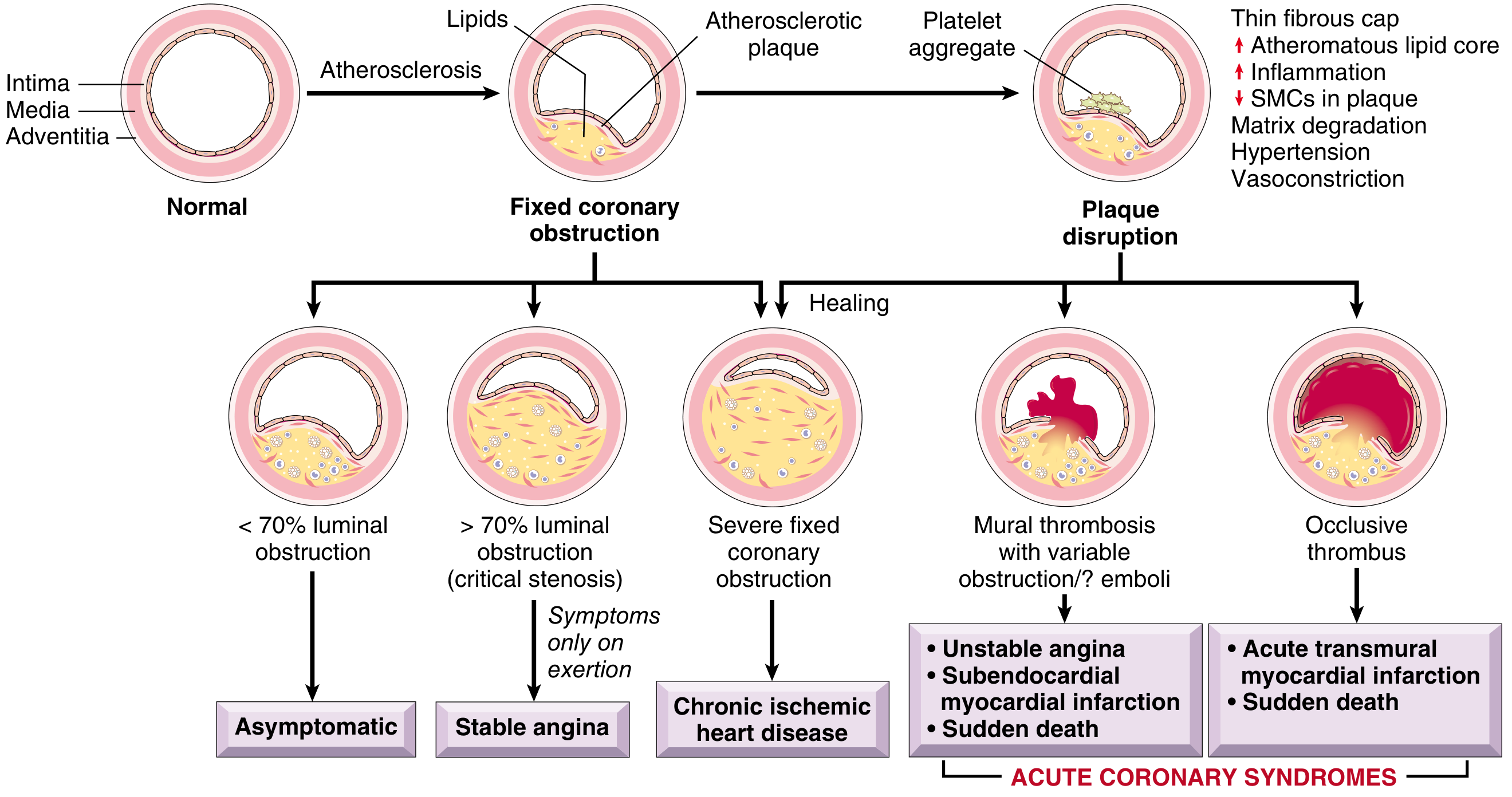
Sequential Steps — Written Summary (Robbins)
① Risk Factors → Endothelial Injury
② Atherosclerotic Plaque Formation
③ Fixed Coronary Obstruction (Chronic)
| Stenosis | Consequence |
|---|---|
| <70% luminal area | Asymptomatic |
| >70% (critical stenosis) | Stable angina — symptoms only on exertion |
| >90% | Ischemia even at rest |
| Slow progressive | Collateral formation may develop |
④ Vulnerable Plaque (the pivot point)
- Large atheromatous lipid core
- Thin fibrous cap
- ↑ Macrophages → metalloprotease secretion → collagen degradation
- ↓ SMCs → reduced collagen synthesis
- Cap thinnest at the shoulder (junction with normal arterial wall) — highest mechanical stress
- External triggers: hypertension, platelet reactivity, vasoconstriction
Robbins key fact: 2/3 of culprit plaques are ≤50% stenotic before rupture; 85% show ≤70% stenosis — i.e., most ACS events arise from previously non-obstructive, asymptomatic plaques.
⑤ Acute Plaque Disruption (Two Mechanisms)
| Plaque Rupture | Plaque Erosion |
|---|---|
| Mechanical stress fractures the fibrous cap | Endothelial cell apoptosis/injury |
| Exposes lipid core & collagen | Exposes subendothelial basement membrane |
| Related to HTN, platelet reactivity | Related to inflammatory & toxic exposures |
⑥ Exposure of Thrombogenic Contents
⑦ Platelet Adhesion & Activation
⑧ Coagulation Cascade → Stable Thrombus
⑨ Thrombus Outcomes → ACS Spectrum
THROMBUS
│
┌──────────────┼──────────────────┐
▼ ▼ ▼
Mural / non- Partial + spasm Complete occlusion
occlusive
± microemboli
│ │ │
▼ ▼ ▼
• Unstable Subendocardial • STEMI
Angina infarct (Transmural MI)
• NSTEMI (inner 1/3) • Sudden
• Subendo- cardiac death
cardial infarct
└──────────────┴──────────────────┘
▼
■ ACUTE CORONARY SYNDROMES ■