Panhypopituitarism
Reading File
Finding Sources
Finding Sources
Searching PubMed
panhypopituitarism hypopituitarism
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have everything needed to compose a thorough answer. Here is a complete, authoritative review of panhypopituitarism:
Panhypopituitarism
Panhypopituitarism refers to a defect in all pituitary hormone lines - a complete failure of anterior pituitary secretion resulting in deficiency of GH, ACTH, TSH, LH, FSH, and prolactin. Posterior pituitary (ADH, oxytocin) may or may not be affected depending on the cause. - Goldman-Cecil Medicine, p. 2399
Hypothalamic-Pituitary Anatomy (Relevant to Pathophysiology)
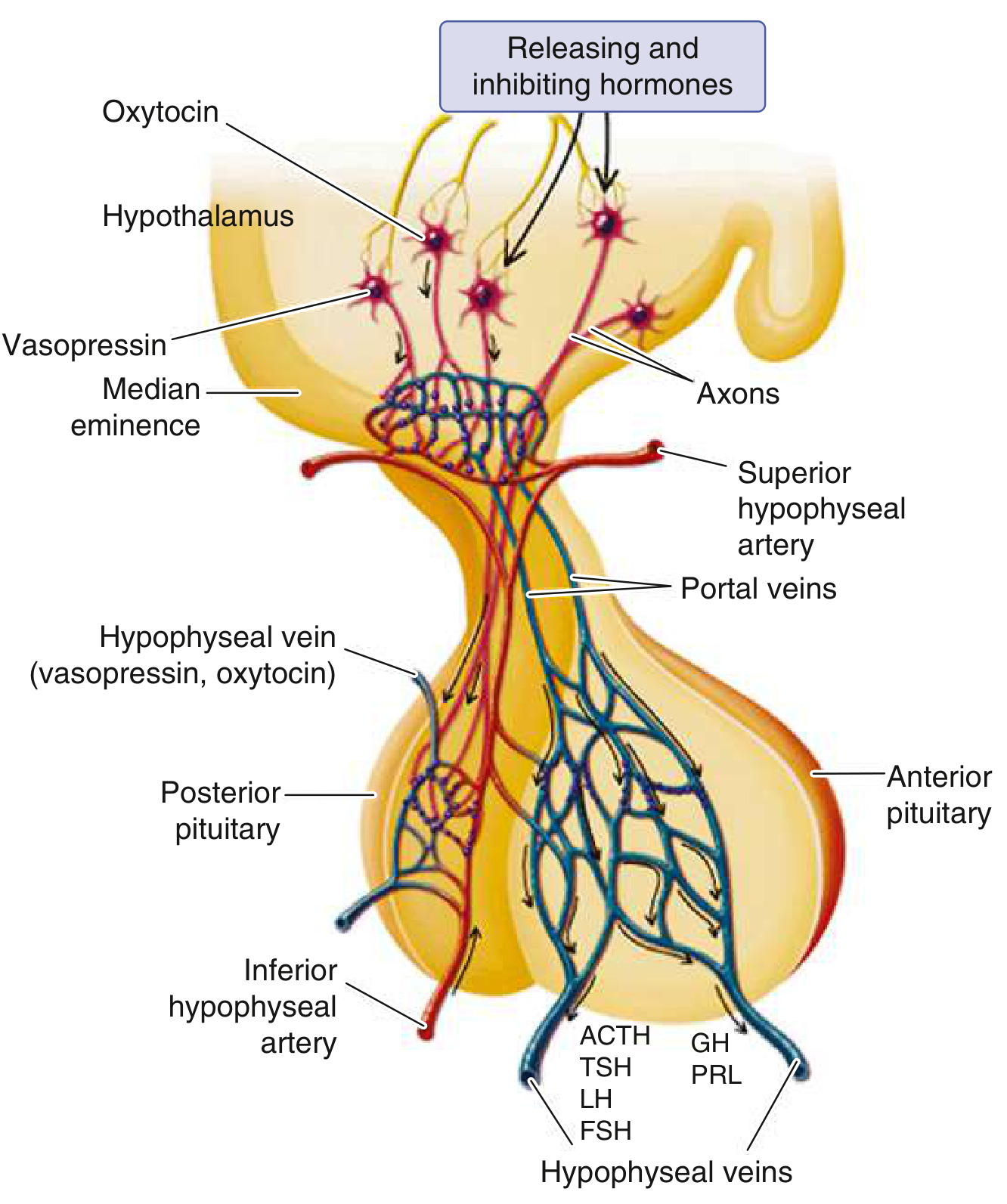
The anterior pituitary receives hypothalamic releasing hormones (GHRH, CRH, TRH, GnRH) via the hypophyseal portal system at the median eminence. The posterior pituitary receives vasopressin and oxytocin axons directly. Hypofunction occurs when approximately 75% of the parenchyma is lost or absent. - Robbins Pathologic Basis of Disease
Etiology and Classification
Congenital / Developmental Causes
| Category | Examples |
|---|---|
| Embryopathic | Anencephaly, pituitary aplasia, septo-optic dysplasia (midline cleft defects) |
| Genetic - transcription factors | PROP1 mutation (most common): GH + PRL + TSH + gonadotropin deficiency; PIT1/POU1F1: GH + PRL + TSH; TPIT: ACTH deficiency; NR5A1/SF1: gonadotrope + adrenal/gonadal |
| Genetic - signaling | HESX1, SOX2, SOX3, LHX3, LHX4, OTX, GLI2, PAX6, BMP4, FGFR1, CHD7, and >50 others |
| Isolated GnRH deficiency | Kallmann syndrome (KAL gene - GnRH deficiency + anosmia) |
Over 80% of PROP1 mutation patients have growth retardation; by adulthood, all are TSH/gonadotropin deficient; a minority later develop ACTH deficiency. - Harrison's 22e, p. 1647
Acquired Causes
Tumors / Mass Lesions
- Pituitary macroadenomas (most common acquired cause) - compress normal cells within the bony sella
- Craniopharyngiomas, meningiomas, gliomas, dysgerminomas
- Metastatic tumors (breast, lung, colon carcinoma), Rathke cleft cysts, hypothalamic hamartomas, lymphoma/leukemia
Vascular
- Sheehan syndrome (postpartum necrosis): most common cause of ischemic necrosis - the anterior pituitary doubles in size during pregnancy without proportional increase in blood supply; postpartum hemorrhage/shock precipitates infarction. The posterior pituitary (direct arterial supply) is typically spared.
- Pituitary apoplexy: sudden hemorrhage into a pituitary gland or adenoma - presents with severe headache, diplopia, acute hypopituitarism; ACTH loss causes acute adrenal insufficiency, hypotension, cardiovascular collapse - a neurosurgical emergency
- Subarachnoid hemorrhage, sickle cell disease, arteritis, snake bite venom, internal carotid aneurysm
Traumatic
- TBI, surgical excision, subarachnoid hemorrhage, contact sports, explosive injury - 25-40% of these patients develop hypothalamic or pituitary dysfunction on long-term follow-up
Radiation
- Up to 2/3 of patients develop hormone insufficiency after ~50 Gy to skull base; occurs over 5-15 years; usually reflects hypothalamic damage rather than direct pituitary destruction. Order of loss: GH > gonadotropins > TSH > ACTH
Infiltrative / Inflammatory
- Sarcoidosis, histiocytosis X, hemochromatosis, amyloidosis, tuberculosis, lymphocytic hypophysitis, granulomatous hypophysitis
- Immune checkpoint inhibitors (CTLA-4 inhibitors, PD-1/PD-L1 inhibitors) - autoimmune hypophysitis
Empty Sella Syndrome
-
Primary: defect in diaphragma sellae allows CSF herniation, compressing pituitary
-
Secondary: mass enlarges sella, then is removed or infarcted
-
Goldman-Cecil Medicine, Table 205-1; Robbins Pathologic Basis of Disease, pp. 821-847; Harrison's 22e, pp. 1638-1793
Sequence of Hormone Loss
A key clinical pattern: trophic hormone failure follows a predictable sequence as pituitary compression/destruction worsens:
GH → FSH → LH → TSH → ACTH
During childhood, growth retardation is usually the first sign; in adults, hypogonadism (secondary amenorrhea or male impotence) is the earliest symptom. - Harrison's 22e, p. 1760
Clinical Features by Hormone Deficiency
| Hormone Lost | Adult Manifestations | Childhood Manifestations |
|---|---|---|
| GH | Decreased lean mass, increased fat, fatigue, dyslipidemia | Growth failure, short stature (pituitary dwarfism) |
| FSH/LH (Gonadotropins) | Females: amenorrhea, infertility, loss of secondary sex characteristics; Males: impotence, loss of libido, loss of pubic/axillary hair | Failure to enter puberty; no adult sexual development |
| TSH | Features of secondary hypothyroidism: lethargy, weight gain, cold intolerance | Growth retardation |
| ACTH | Secondary adrenal insufficiency: hypotension, fatigue, hypoglycemia, hyponatremia (no hyperkalemia - aldosterone preserved) | |
| Prolactin | Failure of postpartum lactation |
The overall picture of adult panhypopituitarism is a lethargic patient who is gaining weight (due to loss of thyroid, GH, ACTH, and adrenocortical hormones) with lost sexual function. - Guyton & Hall Medical Physiology, p. 925
Pituitary dwarfism (childhood panhypopituitarism): proportionate short stature; at age 10 may have the body of a 4-5 year old; at age 20, the body of a 7-10 year old. In 1/3 of dwarfism cases, only GH is deficient - these patients do mature sexually. - Guyton & Hall, p. 925
Sheehan syndrome: classic presentation is failure to lactate postpartum, followed by loss of pubic/axillary hair, then amenorrhea, hypothyroid features, and eventually adrenal insufficiency.
Diagnosis
Biochemical Evaluation
| Hormone | Test | Normal Response |
|---|---|---|
| GH | Insulin tolerance test (ITT): insulin 0.05-0.15 U/kg IV | If glucose <40 mg/dL, GH should rise to >5 µg/L |
| Arginine-GHRH test | GH >4.1 µg/L | |
| Glucagon stimulation test | GH >3 µg/L | |
| ACTH/Cortisol | ITT (gold standard) | Cortisol should rise by >7 µg/dL or reach >20 µg/dL |
| CRH test | ACTH increases 2-4 fold; cortisol reaches 20-25 µg/dL | |
| Short Synacthen (ACTH₂₄) test | Cortisol >18-20 µg/dL at 30-60 min | |
| TSH | Basal TSH + free T4 | Low/normal TSH with low free T4 = secondary hypothyroidism |
| LH/FSH | Basal levels + sex steroids | Low gonadotropins with low testosterone/estradiol |
| Prolactin | Basal serum PRL | Low in panhypopituitarism (contrast with hyperprolactinemia from stalk compression) |
- Goldman-Cecil Medicine, Table 205-2, p. 2400
Basal hormone levels are often insufficient alone; dynamic stimulation testing is required for GH and ACTH.
Imaging
- MRI of the pituitary is the preferred modality - evaluates for mass lesions, empty sella, pituitary size, stalk, and hypothalamic involvement.
Treatment
Two-pronged approach: (1) replace deficient hormones, (2) address the underlying cause. - Goldman-Cecil Medicine, p. 2400
| Deficiency | Replacement |
|---|---|
| ACTH / Adrenal | Hydrocortisone (15-20 mg/day in divided doses, or ~10 mg morning + 5 mg afternoon); stress dosing essential. This is the most life-threatening deficiency - treat first. |
| TSH / Thyroid | Levothyroxine; do NOT replace without first correcting cortisol deficiency (risk of adrenal crisis) |
| LH/FSH / Gonadal | Males: testosterone (IM, transdermal, or gel); Females: estrogen ± progesterone; if fertility desired, gonadotropin injections (FSH + LH or hMG) |
| GH | Recombinant human GH (rhGH) subcutaneous injection; can cure GH-deficient dwarfism if started early |
| ADH (if posterior pituitary involved) | Desmopressin (DDAVP) intranasal or oral |
| Prolactin | No replacement needed in most cases |
Critical teaching point: Always replace glucocorticoids before thyroid hormone - thyroid replacement in the setting of adrenal insufficiency can precipitate a life-threatening adrenal crisis by increasing cortisol metabolism.
Special Situations
- Pituitary apoplexy: neurosurgical emergency; urgent glucocorticoid replacement + consideration of surgical decompression
- Checkpoint inhibitor-induced hypophysitis: a growing cause in oncology patients on CTLA-4 or PD-1/PD-L1 inhibitors; often presents with headache and pituitary enlargement on MRI
- Pregnancy: women with hypopituitarism can achieve pregnancy with gonadotropin therapy but require careful hormonal monitoring throughout
Recent Evidence (PubMed, 2024-2026)
- A systematic review on pituitary stalk sacrifice during craniopharyngioma surgery (Bobeff et al., 2025) confirmed that pituitary stalk preservation significantly reduces postoperative endocrine dysfunction, though recurrence rates must be balanced.
- A meta-analysis on stereotactic radiosurgery for non-functioning pituitary adenomas (De Nigris Vasconcellos et al., 2024) found significant rates of post-SRS hypopituitarism, reinforcing long-term endocrine follow-up after radiation.
- A 2025 report with systematic review identified biallelic POMC mutations as a rare cause of combined pituitary hormonal deficiency associated with severe obesity.
Sources: Guyton & Hall Medical Physiology, p. 924-925 | Robbins Pathologic Basis of Disease, pp. 821-859 | Harrison's Principles of Internal Medicine 22e, pp. 1638-1793 | Goldman-Cecil Medicine, pp. 2398-2401
Wysolone
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
I now have excellent content from multiple authoritative sources. Here is the complete profile of Wysolone (prednisolone):
Wysolone (Prednisolone)
Brand name: Wysolone (Sun Pharma, India)
Generic name: Prednisolone
Class: Synthetic glucocorticoid (corticosteroid)
Available strengths: 5 mg, 10 mg, 20 mg, 40 mg tablets
What Is It?
Prednisolone is the active form of prednisone. Unlike prednisone, which requires hepatic conversion to its active metabolite, prednisolone is directly active - making it the preferred choice in patients with liver disease. It is a synthetic analog of cortisol (hydrocortisone) with predominantly glucocorticoid activity and minimal mineralocorticoid effect.
Mechanism of Action
Corticosteroids exert a pluripotent anti-inflammatory effect via inhibition of inflammatory mediator gene transcription. Specifically:
-
Binds to cytosolic glucocorticoid receptors (GR), causing receptor-ligand complex to translocate to the nucleus
-
Transrepression: Blocks transcription factors NF-κB and AP-1, reducing synthesis of pro-inflammatory cytokines (IL-1, IL-2, IL-6, TNF-α), COX-2, and phospholipase A2
-
Transactivation: Upregulates anti-inflammatory genes (lipocortin/annexin-1, which inhibits phospholipase A2 and arachidonic acid cascade)
-
Reduces vascular permeability, inhibits leukocyte migration, and suppresses T-cell and B-cell function at higher doses
-
Washington Manual of Medical Therapeutics, p. 975; Goodman & Gilman's Pharmacological Basis of Therapeutics
Relative Anti-inflammatory Potency (Comparison Table)
| Drug | Relative Anti-inflammatory Potency | Mineralocorticoid Activity | Biological Half-life |
|---|---|---|---|
| Cortisone | 0.8 | ++ | 8-12 h |
| Hydrocortisone | 1 (reference) | ++ | 8-12 h |
| Prednisolone | 4 | + | 12-36 h |
| Prednisone | 4 | + | 12-36 h |
| Methylprednisolone | 5 | Minimal | 12-36 h |
| Dexamethasone | 25 | None | 36-54 h |
Prednisolone has ~4x the anti-inflammatory potency of hydrocortisone with significantly less mineralocorticoid effect (no significant sodium/water retention at usual doses). - Washington Manual, p. 975
Therapeutic Uses
Wysolone/prednisolone is used across a very wide spectrum:
Rheumatological / Autoimmune
- Rheumatoid arthritis (low-dose, bridging therapy)
- Systemic lupus erythematosus (SLE)
- Polymyalgia rheumatica, vasculitides
- Inflammatory myopathies (dermatomyositis, polymyositis)
- Sarcoidosis
Respiratory
- Acute severe asthma (systemic burst)
- COPD exacerbations
- Interstitial lung disease
- Croup (single dose)
Dermatological
- Severe eczema/atopic dermatitis
- Pemphigus, pemphigoid
- Severe urticaria/angioedema
Gastroenterological
- Inflammatory bowel disease (Crohn's disease, ulcerative colitis) - induction of remission
- Autoimmune hepatitis (combination with azathioprine)
Haematological / Oncological
- Immune thrombocytopenic purpura (ITP)
- Autoimmune hemolytic anemia
- Lymphoma/leukemia regimens (e.g., CHOP, MOPP)
- Nephrotic syndrome (minimal change disease - first-line)
Neurological
- Bell's palsy
- Multiple sclerosis relapses
- Cerebral edema (dexamethasone preferred but prednisolone used)
Endocrine / Replacement
- Congenital adrenal hyperplasia (CAH)
- Addison's disease (less preferred than hydrocortisone)
Transplant
-
Part of immunosuppression regimens post-organ transplant
-
Goodman & Gilman's: "Across the spectrum of inflammatory disease"
Dosing
Dosing is highly disease-dependent:
| Indication | Typical Dose |
|---|---|
| Anti-inflammatory (low dose) | 5-10 mg/day |
| Moderate inflammatory disease | 20-40 mg/day |
| High dose (nephrotic, severe asthma) | 1 mg/kg/day (up to 60-80 mg/day) |
| Pulse / very high dose | 500 mg-1 g IV methylprednisolone preferred |
| Tapering | Gradual reduction once disease controlled |
Key principle: Use the minimum effective dose for the shortest duration possible. - Washington Manual, p. 975
Prednisone vs. prednisolone equivalence: They are equipotent (4x hydrocortisone). 5 mg prednisolone = 5 mg prednisone = 20 mg hydrocortisone = 4 mg methylprednisolone = 0.8 mg dexamethasone.
Adverse Effects
Adverse effects are dose- and duration-dependent. They generally appear at doses >10 mg/day prednisolone equivalent and become significant with prolonged use.
Endocrine / Metabolic
- Hyperglycemia / steroid-induced diabetes
- Weight gain, central obesity, moon face, buffalo hump (iatrogenic Cushing syndrome)
- HPA axis suppression: adrenal suppression can be assumed in patients receiving >20 mg/day for >3 weeks; abrupt discontinuation risks adrenal crisis
- Osteoporosis: start calcium (1-1.5 g/day) + vitamin D (1000 IU/day) from initiation; bisphosphonates for prophylaxis in long-term use
- Growth retardation in children
Cardiovascular
- Dyslipidemia (raised LDL, lowered HDL)
- Hypertension (sodium retention, increased cardiac output)
- Increased risk of atherosclerosis with long-term use
Ophthalmological
- Posterior subcapsular cataracts (dose and duration related)
- Glaucoma (raised intraocular pressure)
Immunological
- Reduced resistance to infections - minor infections can become systemic; quiescent infections (TB, fungi) reactivated
- Infection signs may be partially masked
- Consider PCP (Pneumocystis jirovecii) prophylaxis (cotrimoxazole) for patients on ≥20 mg/day for >1 month
- Reactivation of latent TB - screen with TST/IGRA before starting
Psychiatric
- Insomnia (especially early in therapy)
- Euphoria, emotional lability
- Severe depression or steroid psychosis (at high doses)
Musculoskeletal
- Proximal myopathy (painless weakness of proximal muscles; normal CK/EMG; resolves on stopping)
- Avascular necrosis (osteonecrosis) of femoral head, humeral head, tibial plateau - a serious complication
Dermatological
- Acne, skin atrophy, striae, purpura, easy bruising, poor wound healing
Gastrointestinal
-
GI irritation, peptic ulceration (risk increased with NSAIDs co-prescription)
-
Use PPI prophylaxis if also on NSAIDs or high dose
-
Goodman & Gilman's: "With high-dose/long-term therapy: psychosis, increased susceptibility to infection, osteoporosis, osteonecrosis, myopathy, HPA axis suppression"
-
Washington Manual of Medical Therapeutics, pp. 975-976
-
Yamada's Gastroenterology: "Up to 80% of patients suffer from prednisone/prednisolone-specific side effects"
Key Precautions and Contraindications
| Situation | Consideration |
|---|---|
| Active infections | Caution - may mask signs; treat infection alongside steroids when life-threatening inflammation present |
| Diabetes mellitus | Monitor blood glucose closely; may need insulin adjustment |
| Hypertension | Monitor BP; sodium restriction |
| Peptic ulcer disease | Add PPI; use lowest dose |
| Osteoporosis | Prophylaxis with calcium/vit D/bisphosphonates mandatory |
| Latent TB | Screen and give isoniazid prophylaxis |
| Liver disease | Prednisolone preferred over prednisone (prednisone requires hepatic conversion) |
| Pregnancy | Category C; used when benefits outweigh risks; minimal placental transfer compared to dexamethasone/betamethasone |
| Abrupt cessation | Never stop abruptly after >3 weeks - risk of adrenal crisis; taper gradually |
Tapering
After prolonged use, the dose must be tapered gradually to:
- Allow the HPA axis to recover (prevent adrenal crisis)
- Prevent flare of the underlying disease
A typical taper reduces the dose by ~10-20% every 1-2 weeks, slowing as the dose approaches physiological levels (5-7.5 mg/day equivalent).
Stress Dosing
Patients on long-term prednisolone who undergo surgery, major illness, or severe stress require "stress-dose" glucocorticoids (equivalent to hydrocortisone 50-100 mg IV q6-8h) because their HPA axis cannot mount an adequate cortisol response independently.
Drug Interactions
| Interacting Drug | Effect |
|---|---|
| NSAIDs | Additive GI ulceration risk |
| Antidiabetic drugs | Reduced efficacy (steroid-induced hyperglycemia) |
| Antihypertensives | Reduced efficacy |
| Vaccines (live) | Avoid live vaccines (MMR, varicella) in patients on immunosuppressive doses |
| CYP3A4 inducers (rifampicin, phenytoin) | Reduced prednisolone levels |
| CYP3A4 inhibitors (ketoconazole) | Increased prednisolone levels |
| Warfarin | Variable interaction; monitor INR |
Sources: Washington Manual of Medical Therapeutics, pp. 975-976 | Goodman & Gilman's Pharmacological Basis of Therapeutics | Sleisenger & Fordtran's GI and Liver Disease | Yamada's Textbook of Gastroenterology
This is a shared conversation. Sign in to Orris to start your own chat.