Teach me pemphigus vulgaris, bullous pemphigoid,
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
I now have everything needed for a comprehensive, image-rich teaching answer. Let me compile it.
Autoimmune Blistering Diseases: Pemphigus Vulgaris & Bullous Pemphigoid
These are the two most important autoimmune blistering disorders. Understanding them together - by contrast and comparison - is the best way to master both.
The Big Picture: How to Think About These Diseases
Both are IgG-mediated autoimmune diseases that destroy skin adhesion. The key difference is where the autoantibody attacks:
| Pemphigus Vulgaris | Bullous Pemphigoid | |
|---|---|---|
| Target | Desmoglein (desmosome) | BPAG2/BP180 (hemidesmosome) |
| Level of split | Intraepidermal (suprabasal) | Subepidermal |
| Blister quality | Flaccid, fragile, ruptures easily | Tense, dome-shaped, intact |
| Mucosal involvement | Almost always (often the first sign) | Rare (~20%) |
| Nikolsky sign | Positive | Negative |
| Age group | Middle-aged (40-60s) | Elderly (65-75 years) |
| Prognosis | More serious, potentially fatal | Self-limiting, remitting/relapsing |
The diagram below shows exactly why - the target antigens are at completely different levels of the skin:
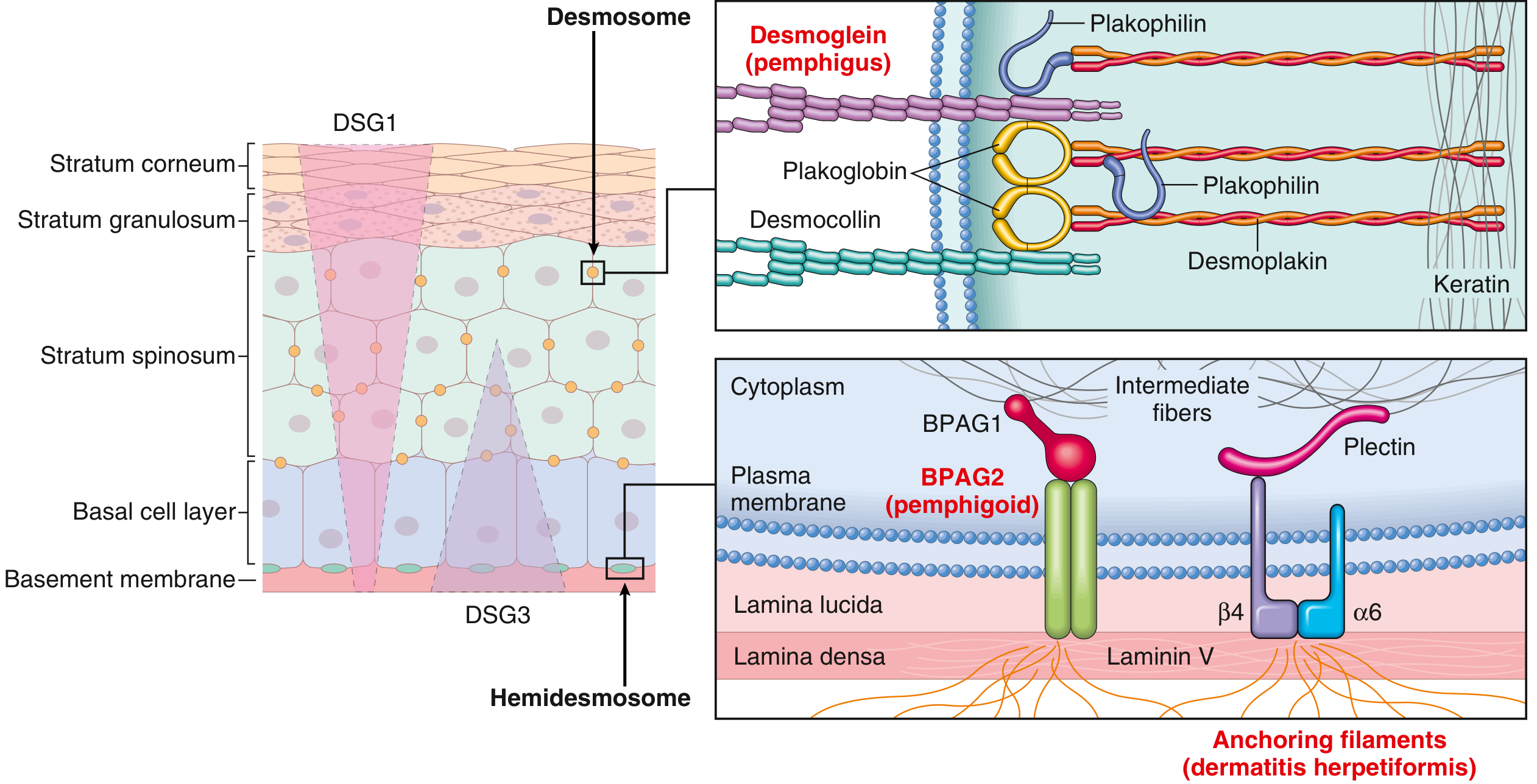
PART 1: PEMPHIGUS VULGARIS (PV)
Epidemiology
- Age of onset: typically 40-60 years
- No sex predilection
- Higher incidence in Ashkenazi Jews and Mediterranean populations
- Associated with HLA-DR4 and HLA-DQ3
Pathogenesis
The fundamental defect is loss of keratinocyte-to-keratinocyte adhesion (acantholysis) caused by IgG autoantibodies against desmoglein-3 (DSG3) and/or desmoglein-1 (DSG1) - cadherin-type glycoproteins that are the trans-adhesion components of desmosomes.
The desmoglein compensation hypothesis explains the clinical distribution of blisters:
- DSG3 is expressed predominantly in the lower epidermis (and mucosae)
- DSG1 is expressed throughout the epidermis, but more in the upper layers
- Mucosal-dominant PV: anti-DSG3 only - blisters occur in the lower epidermis (suprabasal) where only DSG3 compensates
- Mucocutaneous PV: anti-DSG3 + anti-DSG1 - both skin and mucosa are involved
Mechanism of acantholysis: IgG antibody binding to desmoglein triggers intracellular signaling (activation of src kinase, phospholipase C, protein kinase C cascades), leading to:
- Direct steric interference with the DSG3 trans-adhesive interface
- Desmosome disassembly and keratin tonofilament retraction
- Loss of cell-cell cohesion → intraepidermal blister formation
Clinical Features
Skin Lesions
The primary lesion is a flaccid, thin-walled blister that arises on normal-appearing or erythematous skin - classically sparing the palms and soles. Because these blisters are so fragile, you will most often encounter widespread painful erosions rather than intact bullae.
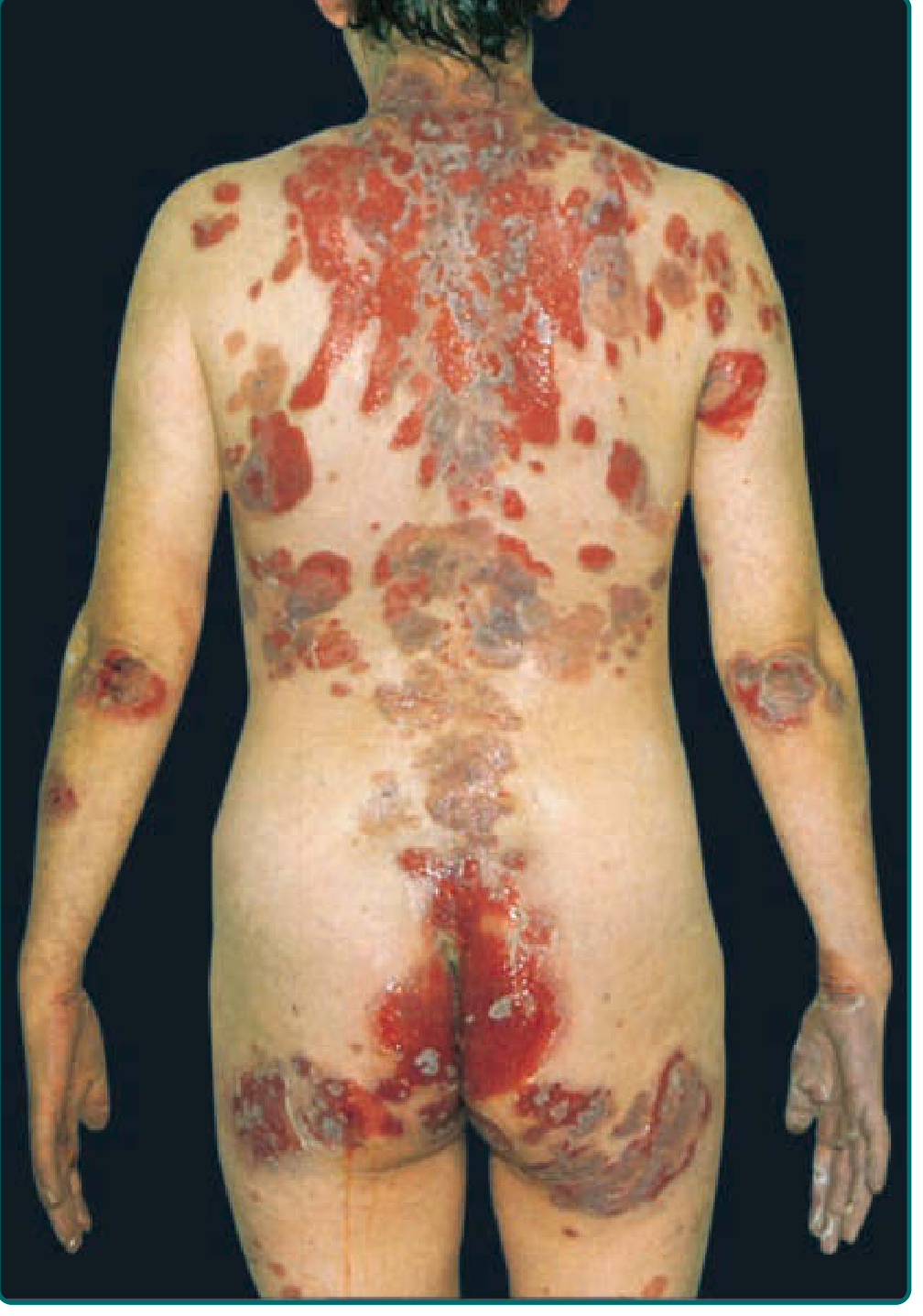
Key features:
- Erosions are large, painful, ooze easily, and bleed
- Tend to spread peripherally rather than heal
- Heal with hyperpigmentation but no scarring
- Pruritus is uncommon (unlike BP)
Pemphigus vegetans is a variant where erosions develop excessive papillomatosis and crusting, especially in intertriginous areas. It has a better prognosis.
Mucous Membrane Lesions
- Oral mucosa is involved in virtually 100% of patients
- Oral involvement typically precedes skin lesions by months (important: most patients are misdiagnosed as aphthous ulcers initially)
- Erosions at buccal and palatine mucosae, ill-defined borders, extremely painful
- Can involve pharynx, esophagus (sloughing casts reported), nasal mucosa, conjunctivae, vagina, penis, and anus
- Intact blisters in the mouth are rare - they rupture before you see them
Nikolsky Sign
Because there is no cohesion within the epidermis, lateral pressure causes the superficial skin layers to slide - this is the Nikolsky sign (positive in PV). Also positive in TEN, SJS, and staphylococcal scalded skin syndrome.
The Asboe-Hansen sign (Nikolsky II) - gentle pressure on an intact bulla spreads fluid laterally under the skin.
Histopathology
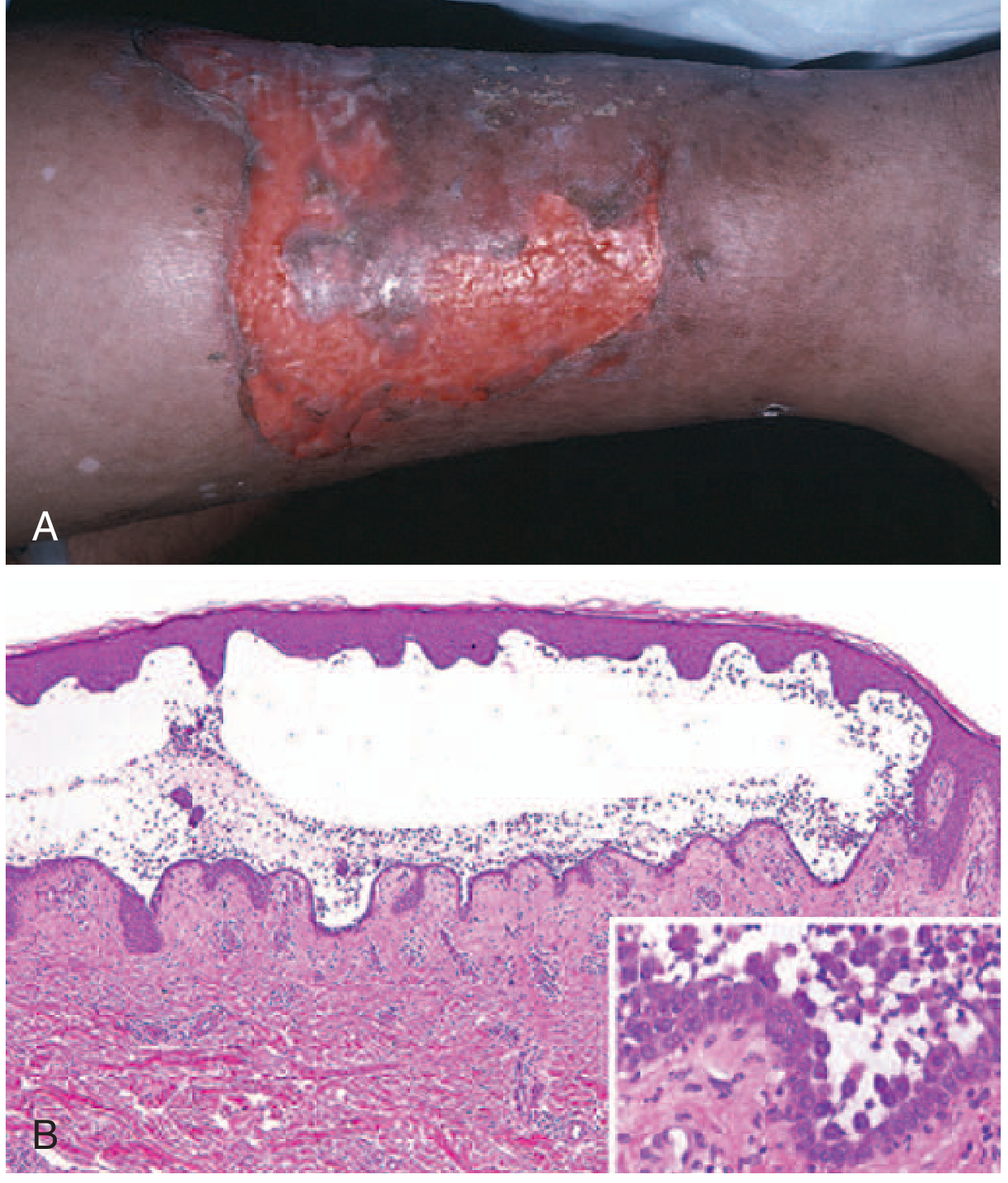
Key histologic findings:
- Suprabasal acantholysis - intraepidermal blister just above the basal cell layer
- The basal cells lose lateral connections but maintain their hemidesmosomal attachment to the basement membrane - classically described as a "row of tombstones"
- A few rounded-up acantholytic keratinocytes float in the blister cavity
- Dermal perivascular mononuclear infiltrate with eosinophils
- No keratinocyte necrosis
Immunofluorescence
The cornerstone of diagnosis:
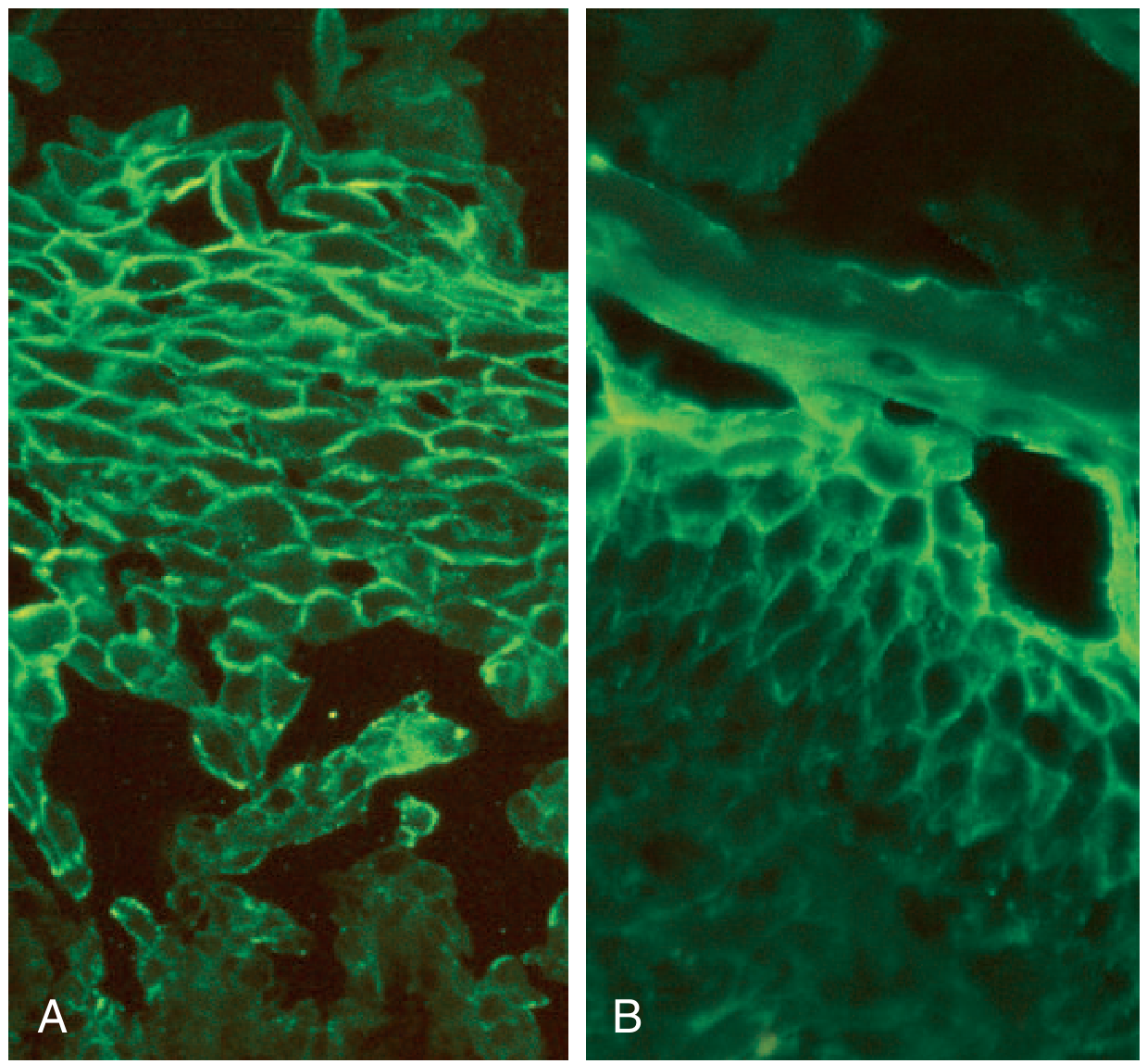
- Direct immunofluorescence (DIF): IgG (and often C3) in an intercellular "fishnet/chicken-wire" pattern throughout the epidermis - biopsy from perilesional skin
- Indirect immunofluorescence (IIF): Circulating anti-DSG antibodies detectable in serum
- ELISA: Anti-DSG1 and anti-DSG3 levels correlate with disease activity - useful for monitoring
Diagnosis Summary
- Clinical presentation (flaccid blisters, oral erosions, Nikolsky positive)
- Biopsy for H&E (suprabasal acantholysis, tombstoning)
- DIF of perilesional skin (fishnet IgG)
- Serology (anti-DSG1, anti-DSG3 by ELISA)
Treatment
Goal: Suppress autoantibody production (not just local inflammation).
Standard Treatment
| Drug | Dose |
|---|---|
| Oral prednisone | 1 mg/kg/day (~60 mg/day) initial dose |
| Rituximab (anti-CD20) | 375 mg/m² weekly x4 OR 1g then 1g at 2 weeks; may repeat every 3-6 months |
Rituximab has transformed PV management - it targets the B-cells producing pathogenic autoantibodies.
Steroid-Sparing / Aggressive
| Drug | Dose |
|---|---|
| Azathioprine | 2-4 mg/kg/day (100-300 mg/day) |
| Mycophenolate mofetil | 2-3 g/day |
| Cyclophosphamide | 1-3 mg/kg/day |
| High-dose IVIg | 400 mg/kg/day x 5 days, repeat monthly |
| Plasmapheresis | 1-2x/week (adjunct) |
| Methotrexate | 7.5-20 mg/week |
Monitoring
- Serum anti-DSG levels track disease activity
- Taper steroids once new blister formation stops (consolidation phase)
Prognosis
Before corticosteroids, PV had a mortality rate >75%. Today, most patients can achieve remission. Mortality is still significant from complications of immunosuppressive therapy (infection, GI bleeding, osteoporosis) rather than the disease itself.
PART 2: BULLOUS PEMPHIGOID (BP)
Epidemiology
- Most common autoimmune blistering disease in Western countries
- Primarily elderly patients (mean onset 65-75 years)
- Risk factors: neurological disorders (dementia, Parkinson disease), psychiatric illness, bedridden state, chronic polypharmacy
- Drug-induced BP: furosemide, penicillamine, captopril, DPP-4 inhibitors (gliptins)
Pathogenesis
IgG autoantibodies target hemidesmosomal proteins at the dermoepidermal junction (DEJ):
- BPAG2 (BP180, type XVII collagen) - the primary pathogenic antigen; transmembrane collagen that spans the lamina lucida
- BPAG1 (BP230) - intracellular plakin protein of hemidesmosomes
The pathogenic sequence:
- Anti-BPAG2 IgG binds to the NC16A domain of BP180 at the DEJ
- Complement activation at the basement membrane zone
- Recruitment of eosinophils and neutrophils (this is important - BP has prominent eosinophils unlike PV)
- Protease release (MMP-9, elastase) degrades anchoring proteins
- Blister forms at the lamina lucida level (subepidermal) - the entire epidermis lifts off intact
Clinical Features
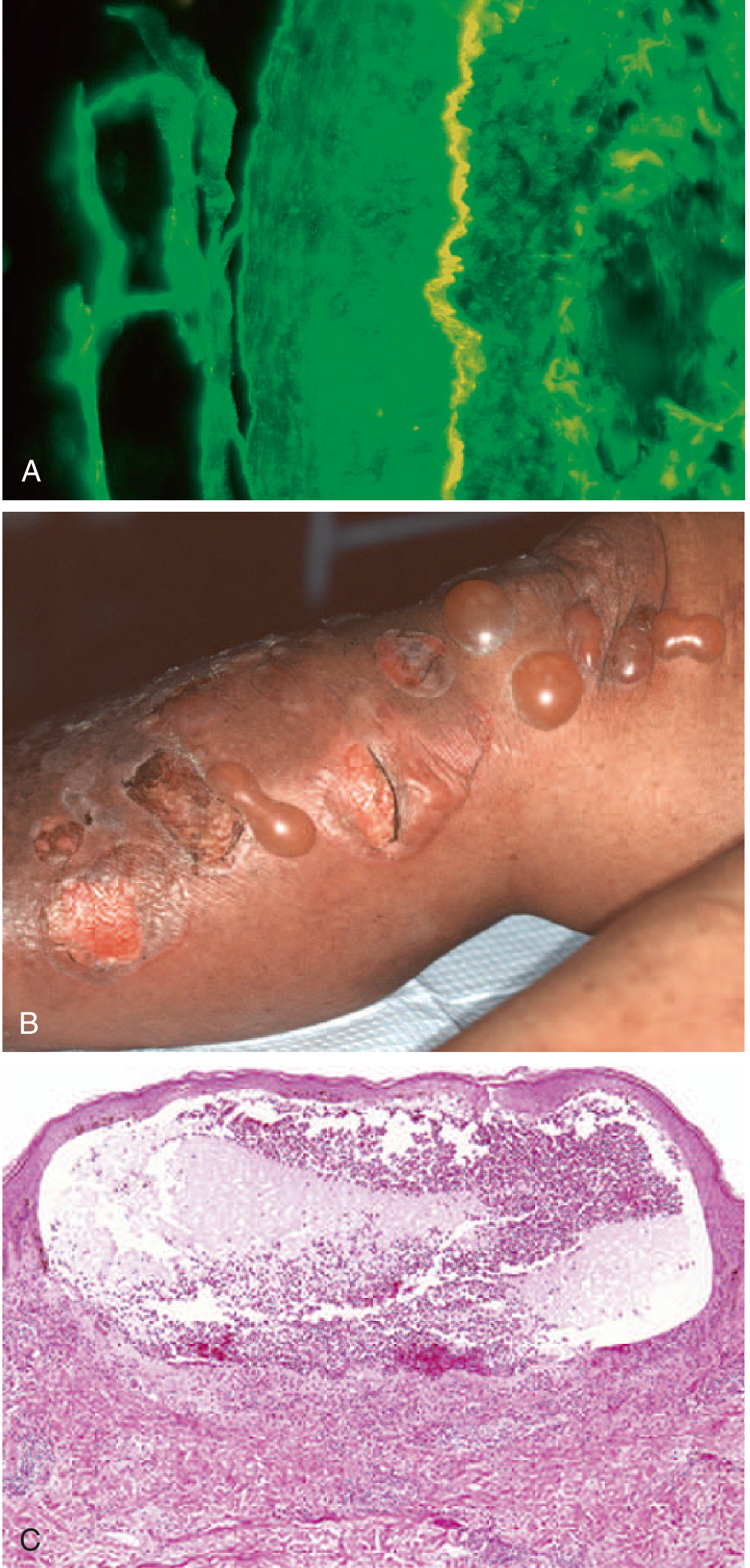
Key: Tense Bullae vs Flaccid Bullae
This is the most important clinical distinction. In BP, the blister forms below the epidermis, so the entire epidermis forms the roof - making bullae tense, dome-shaped, and resistant to rupture.
Clinical course often has two phases:
- Pre-bullous phase (weeks to months): Intense pruritus, erythematous urticarial papules and plaques - no blisters yet. This phase is often misdiagnosed as urticaria or eczema.
- Bullous phase: Large, tense blisters on urticarial/erythematous background
Distribution: Groin, axillae, trunk, thighs, flexor forearms - a flexural predilection (contrast with PV which can be anywhere)
After rupture: Denuded areas that heal spontaneously (do NOT spread like PV erosions) - heal without scarring
Nikolsky sign: NEGATIVE (blisters are subepidermal, epidermis remains anchored)
Pruritus: Very prominent (contrast with PV where pruritus is uncommon)
Mucosae: Involved in ~20% only (much less than PV); pharynx, larynx, eyes rare
Variants to Know
- Gestational pemphigoid (herpes gestationis): Occurs in 2nd-3rd trimester; same BPAG2 antibodies; resolves postpartum, may recur in subsequent pregnancies
- Vesicular pemphigoid: Small, grouped tense blisters (mimics DH)
- Pemphigoid nodularis: Scalp and extremity nodules resembling prurigo nodularis
- Nonbullous variant: Generalized pruritus or urticaria alone - no blisters; difficult to diagnose
Histopathology
- Subepidermal blister - the epidermis lifts off intact as a roof
- No acantholysis (keratinocytes retain intercellular connections - key distinction from pemphigus)
- Prominent eosinophils at the DEJ and within the blister cavity
- Basal cell layer vacuolization in early lesions
- Superficial dermal edema
Immunofluorescence
- DIF: Linear IgG and C3 at the basement membrane zone (linear band pattern along the DEJ) - biopsy from perilesional skin
- IIF: Circulating anti-basement membrane IgG; when salt-split skin is used, antibodies bind to the epidermal side (roof) of the split - this helps distinguish BP from EBA (which binds the dermal floor)
- ELISA: Anti-BP180 ELISA is highly sensitive and specific; correlates with disease activity
Diagnosis Summary
- Clinical: elderly patient, intense pruritus, tense bullae, flexural distribution
- Biopsy H&E: subepidermal blister, no acantholysis, eosinophil-rich
- DIF perilesional: linear IgG + C3 at BMZ
- ELISA: anti-BP180 and anti-BP230
Treatment
Localized BP:
- High-potency topical corticosteroids alone (clobetasol 0.05% BID) - effective even in moderate-severe disease and may be safer than systemic steroids in the elderly
Extensive BP:
- Oral prednisone 0.75-1 mg/kg/day
- After blisters controlled, taper slowly (5 mg/week down to 30 mg, then alternate-day taper)
- Steroid-sparing agents: azathioprine, mycophenolate mofetil, methotrexate
Alternative / Adjunct options:
- Tetracycline (doxycycline) + nicotinamide - effective with fewer steroid side effects; preferred option in trials comparing to prednisone in older patients
- Dapsone - controls 15-44% of BP patients
- Rituximab - for refractory cases
- Omalizumab - used in BP given the IgE-mediated component (eosinophilia, pruritus suggest IgE involvement)
- IVIg, plasmapheresis - severe refractory cases
Important: In the elderly, steroid complications (osteoporosis, diabetes, immunosuppression) can be more dangerous than the disease itself. Minimize corticosteroid exposure.
Prognosis
- Self-limiting in most patients (unlike PV)
- Remitting-relapsing course
- Does not leave scars
- Mortality is associated with disease severity and treatment complications (not the disease itself)
- Children: usually resolves within 5 months
SIDE-BY-SIDE COMPARISON TABLE
| Feature | Pemphigus Vulgaris | Bullous Pemphigoid |
|---|---|---|
| Autoantigen | DSG3 (+/- DSG1) | BPAG2 (BP180) + BPAG1 (BP230) |
| Adhesion structure | Desmosome | Hemidesmosome |
| Blister level | Suprabasal (intraepidermal) | Subepidermal |
| Blister type | Flaccid, ruptures easily | Tense, dome-shaped |
| Nikolsky sign | Positive | Negative |
| Mucous membranes | Nearly 100% | ~20% |
| Pruritus | Uncommon | Prominent (often precedes bullae) |
| Age | 40-60 years | 65-75 years |
| Histology | Suprabasal acantholysis, tombstoning | Subepidermal blister, NO acantholysis, eosinophils |
| DIF pattern | Intercellular "fishnet/chicken-wire" IgG | Linear IgG + C3 at BMZ |
| Salt-split skin IIF | N/A | Epidermal side (roof) |
| Key serology | Anti-DSG1, Anti-DSG3 | Anti-BP180, Anti-BP230 |
| Inflammation | Eosinophils present but modest | Prominent eosinophils |
| Scarring | No | No |
| Prognosis | Serious (pre-Rx fatal) | More benign, self-limiting |
| First-line Rx | Prednisone + Rituximab | Topical clobetasol / Prednisone ± AZA |
MEMORY TRICKS
- Pemphigus = Painful mucosa, Potentially fatal, Placid/flaccid blisters, Positivity for Nikolsky
- Bullous pemphigoid = Big tense Bullae, BMZ target, Benign course, older patient Bodies
- The "V" in Vulgaris - think of the Vital mucosa being involved first
- PV splits the epidermis → roof is thin → fragile; BP splits below the epidermis → roof is full epidermis → tough
- DIF pattern: PV = spider web (intercellular); BP = line (at the basement membrane)
Sources: Fitzpatrick's Dermatology (Vol 1-2), Dermatology 2-Volume Set 5e, Robbins & Kumar Basic Pathology, Andrews' Diseases of the Skin, Robbins Cotran & Kumar Pathologic Basis of Disease
Now teach me lichen planus, psoriasis,
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Loading Image
Reading File
Reading File
I now have all the information needed. Let me compile the comprehensive teaching response.
Lichen Planus & Psoriasis
Both are common, chronic inflammatory skin diseases - but they differ fundamentally in mechanism, morphology, and management. Psoriasis is primarily a T cell-driven hyperproliferative disorder; lichen planus is an autoimmune/interface dermatitis where T cells attack basal keratinocytes.
PART 1: LICHEN PLANUS (LP)
The 6 P's - The Classic Memory Aid
The primary lesions of LP are described by 6 P's:
Pruritic, Planar (flat-topped), Polygonal, Purple (violaceous), Papules and Plaques
These are almost pathognomonic when all 6 are present.
Epidemiology
- Affects ~0.3% of the population worldwide; all races
- Cutaneous LP peaks between ages 40-70 in persons of European descent
- Childhood LP is more common in Asian/Indian/Arab populations (up to 10-20%)
- Associated with HLA-DR/DQ, HLA-A3 alleles
- Oral LP affects women slightly more than men (2.3% vs 1.5%)
- Strong association with hepatitis C virus (HCV) - especially in Mediterranean/Italian populations; screening is warranted
Pathogenesis
LP is a T-cell-mediated autoimmune disease (not primarily antibody-mediated like pemphigus). The mechanism:
- CD8+ T lymphocytes recognize an (unknown) antigen on basal keratinocytes
- T cells cluster at the dermoepidermal junction and launch a cytotoxic attack
- Th1 cytokines (IFN-γ, TNF-α) predominate
- Basal keratinocytes undergo apoptosis → liquefactive degeneration of the basal cell layer (also called hydropic degeneration or vacuolar alteration)
- Dead basal cells become eosinophilic globules = Civatte bodies (colloid bodies)
- The inflammatory infiltrate "hugs" the DEJ like a band = lichenoid reaction pattern
This same histologic pattern ("interface dermatitis with lichenoid infiltrate") is the template for all lichenoid drug reactions and other interface dermatoses.
Clinical Features
Classic Skin Lesions
Small, flat-topped, polygonal papules with a violaceous (purple-pink) color and glistening surface. On close inspection (or dermoscopy), the surface shows fine white lines crossing the lesion - Wickham striae - a nearly pathognomonic finding.
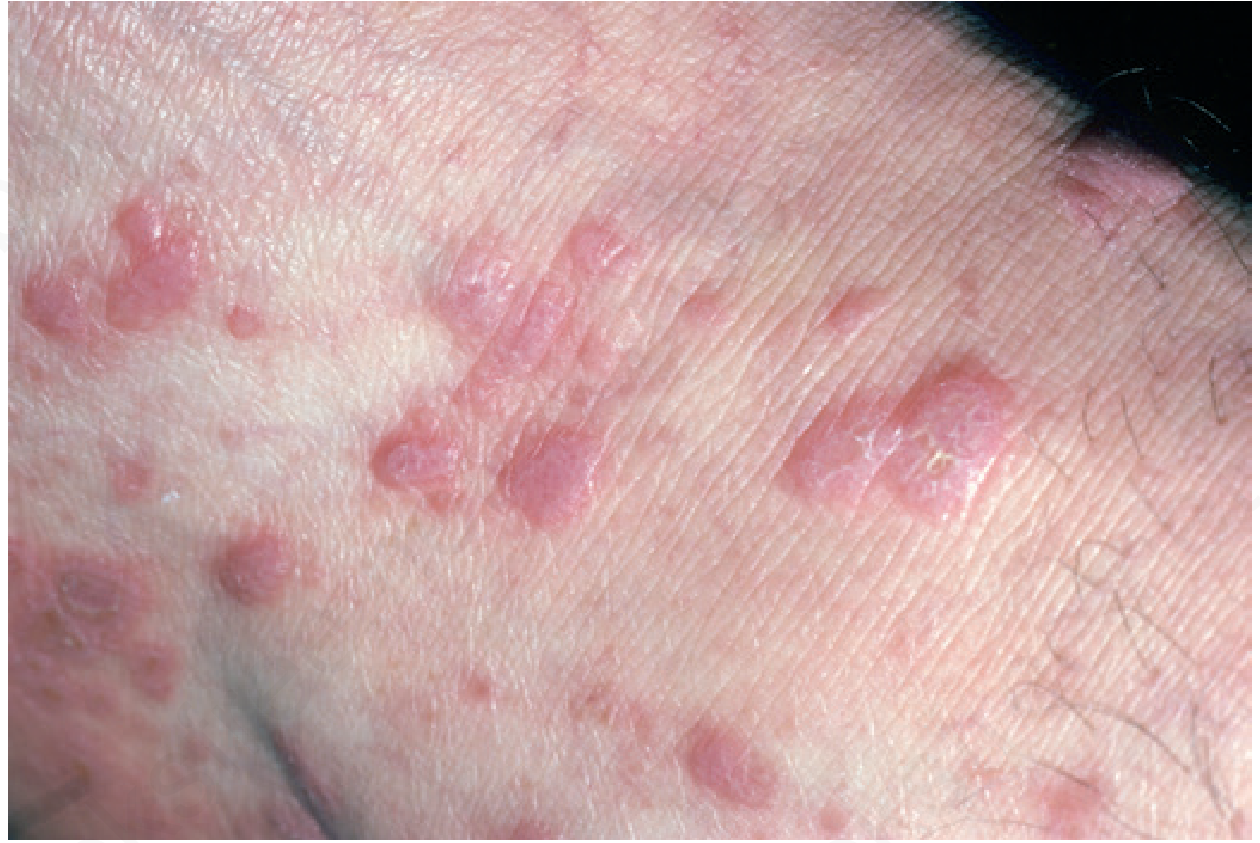
Sites of predilection: Flexor wrists (most classic), trunk, medial thighs, shins, dorsal hands, glans penis. Face is only rarely involved.
Koebner phenomenon: LP lesions appear at sites of skin trauma (scratches, scars).
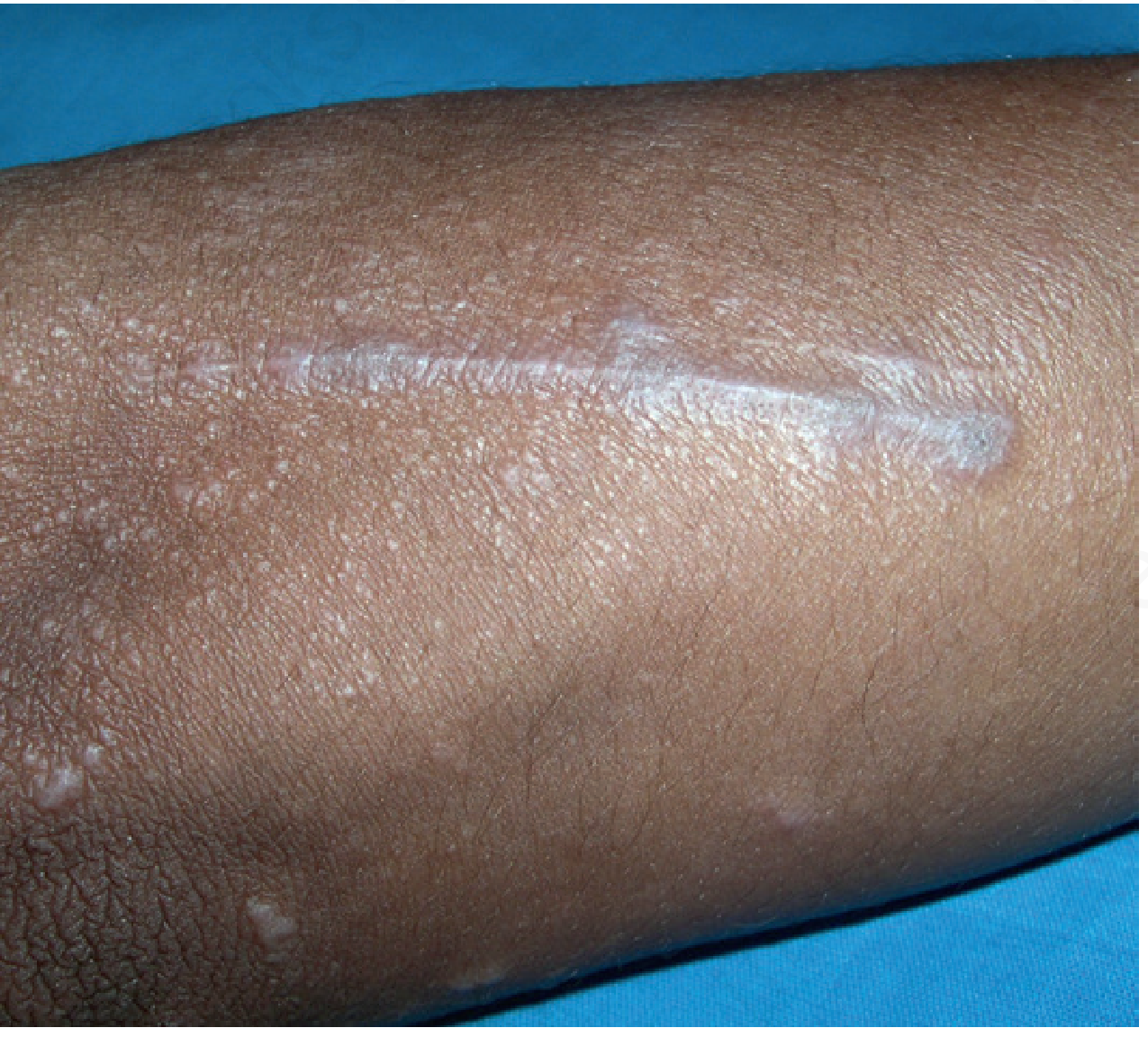
Pruritus is often prominent and may precede visible lesions. Intensity of itch is often disproportionate to the visible disease.
Resolution: Lesions heal with post-inflammatory hyperpigmentation (especially in darker skin types). Two-thirds of patients clear within 1 year spontaneously.
Variants to Know
| Variant | Features |
|---|---|
| Hypertrophic LP | Thick, verrucous plaques; anterior shins; chronic; "igneous rock" appearance |
| Annular LP | Ring-shaped; predilection for penis/scrotum/axilla; mostly asymptomatic |
| Linear LP | Follows Blaschko lines; more common in children |
| Follicular LP (LPP) | Lichen planopilaris - scalp; scarring alopecia |
| Bullous LP | Blisters within LP lesions from severe vacuolar degeneration |
| LP pemphigoides | LP + bullous pemphigoid-like blistering; anti-BP180 antibodies |
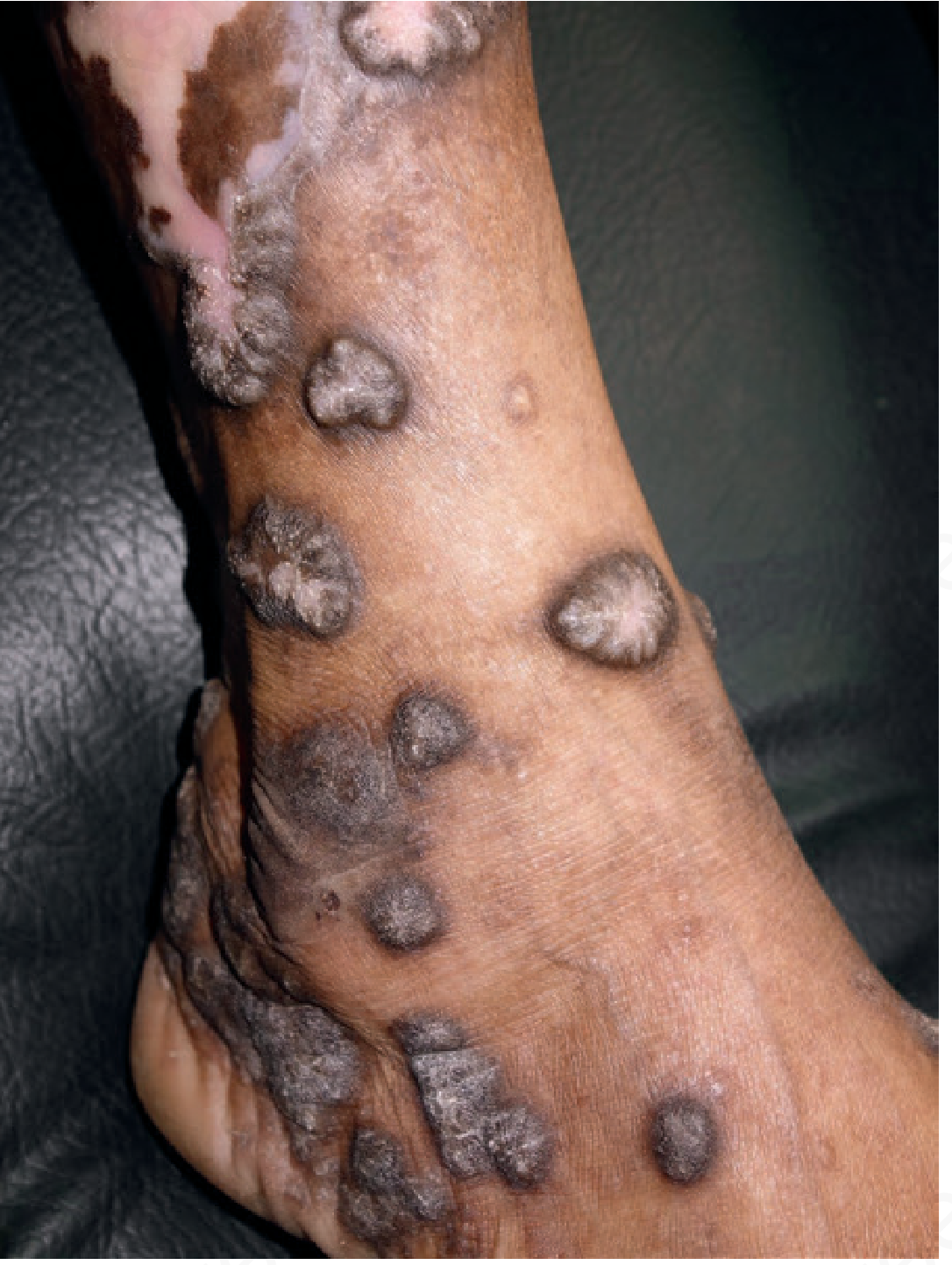
Oral LP
Oral LP is common (affects up to 65% of LP patients) and exists in several forms:
- Reticular (most common): Asymptomatic interlacing white striae (Wickham striae); buccal mucosa; often bilateral
- Erosive: Painful red erosions; higher malignant transformation risk (~1-2% to oral SCC over 10 years)
- Atrophic: Red/white atrophic patches
- Bullous/plaque/papular: Less common variants
Important: Oral LP is associated with HCV (screen patients, especially in high-prevalence regions).
Nail LP (~5-10% of patients)
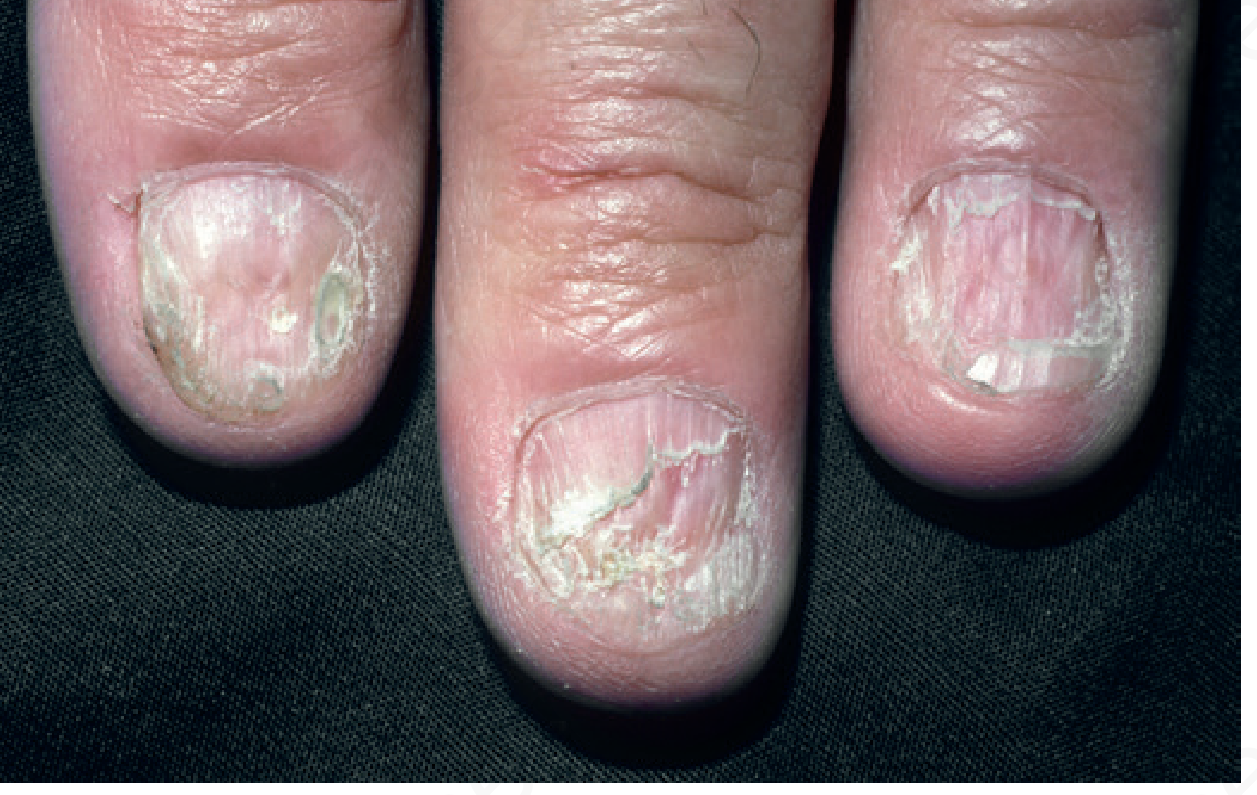
- Longitudinal ridging and splitting (90% of nail LP cases)
- Onycholysis, subungual debris
- Red lunulae (30%)
- Pterygium - pathognomonic: scarring of the nail fold over the nail plate
- Severe disease leads to permanent anonychia (loss of nail plate)
Scalp LP (Lichen Planopilaris)
- Perifollicular erythema and scale
- Leads to permanent scarring alopecia (unlike LP elsewhere which is non-scarring)
- Associated with frontal fibrosing alopecia variant
Histopathology
The classic triad:
- Band-like ("lichenoid") lymphocytic infiltrate at the DEJ - the infiltrate obscures and "hugs" the basement membrane zone
- Vacuolar/hydropic degeneration of the basal cell layer - basal keratinocytes are destroyed
- Civatte bodies (colloid/hyaline bodies) - dead, eosinophilic basal keratinocytes that have undergone apoptosis
Additional features:
- Saw-tooth" (irregular) rete ridges - the inflammatory destruction creates a jagged silhouette
- Hypergranulosis (thickened granular layer) - correlates with Wickham striae clinically
- No significant epidermal hyperplasia (distinguishes from psoriasis)
- DIF: Linear or shaggy fibrinogen deposits at BMZ; IgM in Civatte bodies
Triggers / Associations
- HCV infection (strongest known association, especially erosive oral LP)
- Drugs (lichenoid drug reactions): NSAIDs, beta-blockers, ACE inhibitors, antimalarials, thiazides, gold, penicillamine
- Dental amalgam can trigger oral LP at contact sites
- Hepatitis B vaccination (rare)
- PD-1 inhibitors (pembrolizumab) - checkpoint inhibitor-associated LP
- Stress, trauma (Koebner)
Malignant Potential
Oral erosive LP carries a ~1-2% risk of transformation to oral squamous cell carcinoma over a 10-year period. Hypertrophic LP of the skin also has a small SCC risk. Regular surveillance of erosive oral LP is essential.
Treatment
Therapeutic ladder for LP (from Dermatology 5e):
| Approach | Agents | Evidence |
|---|---|---|
| Topical (first-line) | Superpotent corticosteroids (clobetasol) | Level 1-2 |
| Topical | Tacrolimus, pimecrolimus | Level 1-3 |
| Topical oral | Calcineurin inhibitors (cyclosporine rinse) | Level 1 |
| Phototherapy | Narrowband UVB, PUVA, UVA1, excimer | Level 2 |
| Systemic | Prednisone 15-20 mg/day x 2-6 weeks then taper | Level 1 |
| Systemic | Acitretin (retinoid) | Level 2 |
| Systemic | Hydroxychloroquine | Level 2 |
| Systemic | Methotrexate | Level 2 |
| Systemic | Apremilast (PDE4 inhibitor) | Level 2-3 |
| Refractory | Cyclosporine, JAK inhibitors | Level 2-3 |
Important: Do NOT use systemic steroids long-term for LP - use them for acute control only, then transition to steroid-sparing agents.
PART 2: PSORIASIS
Overview
Psoriasis is one of the most common immune-mediated inflammatory skin diseases, affecting 1-2% of the population worldwide. It is a systemic disease - the skin inflammation is the most visible manifestation, but cardiovascular disease, metabolic syndrome, psoriatic arthritis, and reduced life expectancy are all part of the picture.
Epidemiology
- Equal frequency in men and women
- Bimodal age onset: 16-22 years (type I, HLA-linked, more severe) and 57-60 years (type II)
- Genetics: When both parents have psoriasis, ~50% risk in offspring; one parent affected → ~16%
- HLA-Cw6 is the strongest genetic association (type I psoriasis)
- MHC Class I locus confers the largest genetic risk
- Less common in Sub-Saharan African and Native American populations
Pathogenesis
Psoriasis is a Th1/Th17-driven immune disease causing keratinocyte hyperproliferation. The key cytokine axis:
Trigger → Dendritic cell activation → IL-23 production → Th17 cell differentiation → IL-17A/IL-17F → Keratinocyte activation → Rapid epidermal turnover + inflammatory recruitment
The full cascade:
- An environmental trigger (infection, trauma, drug, stress) activates plasmacytoid and myeloid dendritic cells
- DCs produce TNF-α, IL-12, IL-23
- IL-23 drives Th17 cell development and survival
- Th17 cells produce IL-17A, IL-17F, IL-22
- IL-17 acts on keratinocytes → neutrophil recruitment (IL-8), antimicrobial peptide production
- IL-22 causes acanthosis (thickened epidermis) by retarding keratinocyte differentiation
- Basal keratinocyte transit time falls dramatically (from ~52 days normally to ~8 days) → parakeratosis, poor cornification
- Vascular changes: papillary capillary loops elongate, dilate, and "kiss" the epidermis → "squirting papillae" (neutrophils and lymphocytes extravasate directly at papillary tips) → Auspitz sign
Why this matters for treatment: The IL-23/IL-17 axis is the pharmacological bull's-eye for modern biologics.
Triggers and Exacerbating Factors
| Trigger | Notes |
|---|---|
| Streptococcal pharyngitis | Classically triggers guttate psoriasis in children |
| Koebner phenomenon | Psoriasis appears at sites of skin trauma |
| Stress | Aggravates in ~50% of patients |
| Drugs | β-blockers, lithium, antimalarials, terbinafine, NSAIDs, IFN, IL-2; systemic steroids withdrawal → pustular flare (NEVER use systemic steroids for psoriasis) |
| HIV | Severe, recalcitrant psoriasis |
| Pregnancy | Usually improves during pregnancy, flares postpartum |
Clinical Types
| Type | Features |
|---|---|
| Plaque (vulgaris) | 80-90% of cases; chronic, well-demarcated plaques; elbows, knees, scalp, sacrum |
| Guttate | Small "raindrop" papules, often post-streptococcal; trunk; often resolves spontaneously |
| Inverse | Skin folds (axillae, groin, inframammary); no scale; often misdiagnosed as fungal |
| Pustular | Sterile pustules; localized (palmoplantar) or generalized (von Zumbusch - medical emergency) |
| Erythrodermic | >90% BSA involvement; medical emergency; hypothermia, high-output cardiac failure risk |
| Nail | Pitting, onycholysis, "oil spots" (salmon patches), onychauxis, subungual hyperkeratosis |
| Scalp | Thick scaly plaques extending beyond the hairline; hair loss may occur |
| Psoriatic arthritis | Occurs in ~30% of patients; can occur without skin disease |
Clinical Signs to Know
Auspitz sign: Pinpoint bleeding spots appear when scales are removed from a psoriatic plaque. This is because the elongated papillary capillaries reach close to the surface - scale removal exposes them.
Woronoff ring: A pale ring or halo of blanching surrounding a psoriatic plaque (especially after phototherapy).
Koebner phenomenon: New psoriatic lesions appear at sites of trauma.
"Candle grease" sign: Gentle scraping of a psoriatic plaque produces a stratified silvery-white scale that flakes off like candle wax.
Histopathology
Psoriasis has a distinctive - and important - histologic picture:
| Feature | Significance |
|---|---|
| Acanthosis | Thickened epidermis with long, regular, bulbous rete ridges ("test tube" or "club-shaped") |
| Parakeratosis | Retained nuclei in stratum corneum (rapid turnover = no time for nucleus loss) |
| Absent granular layer | Cells divide too fast to form a granular layer (correlates with parakeratosis) |
| Munro microabscesses | Neutrophils collected in the stratum corneum (within parakeratotic foci) - pathognomonic |
| Spongiform pustules of Kogoj | Neutrophils within the stratum spinosum (more prominent in pustular psoriasis) |
| Dilated tortuous papillary capillaries | "Kissing" capillaries close to the thinned suprapapillary epidermis → Auspitz sign |
| Thinned suprapapillary plate | Epidermis is paradoxically thin directly above dermal papillae |
| Lymphocytic infiltrate | Perivascular and within the epidermis; T cells predominate |
| Minimal spongiosis | Distinguishes psoriasis from eczema/dermatitis |
Key histologic distinction from eczema: Psoriasis has minimal spongiosis + prominent neutrophils in the stratum corneum; eczema has prominent spongiosis and lymphocytes without Munro abscesses.
Nail Psoriasis
Nail involvement occurs in ~50% of skin psoriasis patients (and is a strong predictor of psoriatic arthritis):
- Pitting (most common) - from matrix disease
- Onycholysis - nail separates from nail bed
- "Oil drop" / "salmon patch" sign - yellow-brown discoloration under the nail (pathognomonic)
- Subungual hyperkeratosis
- Leukonychia, red lunulae, splinter hemorrhages
Psoriatic Arthritis
Occurs in ~30% of psoriasis patients. Types:
- Oligoarticular asymmetric (most common)
- Polyarticular (resembles RA)
- Distal interphalangeal (DIP) joint predominant (classic psoriatic pattern)
- Axial (spondylitis/sacroiliitis - HLA-B27 associated)
- Arthritis mutilans (most severe - "opera glass" deformity)
Dactylitis ("sausage digit") - diffuse swelling of entire finger/toe - is a hallmark of psoriatic arthritis.
Systemic Comorbidities
Psoriasis is a systemic inflammatory disease:
- Cardiovascular disease (increased risk of MI, stroke - especially in young patients with severe psoriasis)
- Metabolic syndrome (obesity, hypertension, dyslipidemia, insulin resistance)
- Psoriatic arthritis
- Depression and anxiety
- Inflammatory bowel disease (Crohn's more than UC)
- Slightly increased risk of lymphoma
- Increased risk of celiac disease
Treatment
Topical (Mild-Moderate, Limited BSA)
| Agent | Mechanism | Notes |
|---|---|---|
| Corticosteroids (clobetasol, betamethasone) | Anti-inflammatory | First-line; 68-89% respond to superpotent steroids |
| Calcipotriene (calcipotriol) | Vitamin D analog; inhibits keratinocyte proliferation | Comparable efficacy to potent steroids; combination with steroids is most effective |
| Tazarotene | Retinoid; normalizes keratinocyte differentiation | Slightly more adverse effects; combined with steroids reduces irritation |
| Tacrolimus/pimecrolimus | Calcineurin inhibitors | Best for facial and inverse psoriasis; not thick plaques |
NEVER use systemic corticosteroids for psoriasis - withdrawal triggers life-threatening generalized pustular psoriasis (von Zumbusch).
Phototherapy
- Narrowband UVB (NB-UVB) - first-line phototherapy; safe in pregnancy
- PUVA (psoralen + UVA) - effective but increases long-term skin cancer risk
- Excimer laser - targeted 308nm UVB for localized plaques
Systemic (Moderate-Severe)
| Drug | Mechanism | Key points |
|---|---|---|
| Methotrexate | Folate antagonist; anti-inflammatory | 5-15 mg/week oral; monitor LFTs; teratogenic; effective for both skin and arthritis |
| Acitretin | Retinoid; normalizes differentiation | Good for pustular, palmoplantar; teratogenic (2 years contraception after stopping) |
| Cyclosporine | Calcineurin inhibitor; T-cell suppression | Rapid onset; limit to 1-2 years (nephrotoxicity, hypertension) |
| Apremilast | PDE4 inhibitor | Oral; no monitoring required; modest efficacy |
Biologics - The Modern Cornerstones
Targeting the IL-23/IL-17 axis has transformed psoriasis management:
| Drug | Target | Class | Notes |
|---|---|---|---|
| Etanercept | TNF-α (receptor fusion) | Anti-TNF | First biologic approved for psoriatic arthritis |
| Infliximab | TNF-α (monoclonal Ab) | Anti-TNF | IV; fastest onset among TNF inhibitors |
| Adalimumab | TNF-α (monoclonal Ab) | Anti-TNF | SC; good for skin + arthritis |
| Ustekinumab | IL-12/23 (p40 subunit) | Anti-IL-12/23 | Less frequent dosing (q12 weeks) |
| Secukinumab | IL-17A | Anti-IL-17 | Very high efficacy (PASI 90 in ~60%); rapid onset |
| Ixekizumab | IL-17A | Anti-IL-17 | Similar to secukinumab |
| Guselkumab, risankizumab | IL-23 (p19 subunit) | Anti-IL-23 | High efficacy; very durable responses |
Key teaching point: IL-17 inhibitors (secukinumab, ixekizumab) are among the most effective agents for plaque psoriasis, confirming IL-17 as the central effector cytokine.
COMPARISON TABLE: LP vs PSORIASIS
| Feature | Lichen Planus | Psoriasis |
|---|---|---|
| Mechanism | T cell-mediated interface dermatitis | Th1/Th17 keratinocyte hyperproliferation |
| Primary lesion | Flat-topped, polygonal, violaceous papule | Erythematous plaque with silvery scale |
| Surface | Wickham striae (white lines) | Thick, micaceous (silvery) scale |
| Color | Violaceous/purple | Salmon-pink/red |
| Classic site | Flexor wrists | Extensor surfaces (elbows, knees) |
| Scale | Scant, adherent | Thick, silvery, easily removed |
| Koebner | Yes | Yes |
| Auspitz sign | No | Yes (pinpoint bleeding on scale removal) |
| Mucosal involvement | Common (Wickham striae in mouth) | Unusual |
| Nail | Pterygium, ridging, trachyonychia | Pitting, oil drop, onycholysis |
| Histology | Band-like lymphocytes at DEJ, Civatte bodies, saw-tooth rete, hypergranulosis | Acanthosis, parakeratosis, Munro microabscesses, absent granular layer, dilated papillary capillaries |
| Key cells | CD8+ T cells at DEJ | Th17 cells, neutrophils, keratinocytes |
| Key cytokines | IFN-γ, TNF-α | IL-17, IL-23, TNF-α |
| Prognosis | Usually self-limiting (skin); oral can be chronic | Chronic, relapsing; systemic disease |
| Malignant risk | Oral erosive LP → oral SCC (~1-2%) | Increased lymphoma; no direct SCC risk from plaques |
| First-line Rx | Topical corticosteroids | Topical corticosteroids + calcipotriene |
| Systemic Rx | Prednisone short-course, acitretin | MTX, cyclosporine, acitretin, biologics |
| Biologic targets | Not commonly used | Anti-TNF, anti-IL-17, anti-IL-23 |
Memory Tricks
- LP = "Lacy Purple" - the Wickham striae look like white lace on a purple background
- LP histology = BALL - Band-like infiltrate, Acantholysis absent, Liquefactive degeneration, Lyssa (Civatte) bodies
- Psoriasis = "Silver Scale on Salmon Skin" at extensor surfaces
- Psoriasis histology = PALM - Parakeratosis, Acanthosis, Lost granular layer, Munro microabscesses
- Auspitz sign = A for Auspitz, A for Arteries (the capillaries exposed by scale removal bleed)
- For biologics in psoriasis: TNF → IL-12/23 → IL-23 → IL-17 (progressive refinement of the pathway)
Sources: Andrews' Diseases of the Skin (Clinical Dermatology), Fitzpatrick's Dermatology (Vol 1-2), Textbook of Family Medicine 9e, Dermatology 2-Volume Set 5e
Now teach me systemic sclerosis, DLae, Eczema
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
I now have sufficient content from all three topics. Let me compose the comprehensive teaching response.
Systemic Sclerosis, Discoid Lupus Erythematosus & Atopic Dermatitis (Eczema)
PART 1: SYSTEMIC SCLEROSIS (SSc / Scleroderma)
What It Is in One Sentence
SSc is a chronic autoimmune connective tissue disease defined by a triad: autoimmunity + obliterative vasculopathy + progressive fibrosis, affecting skin and internal organs.
Epidemiology
- Incidence: 9-19 cases per million per year (orphan disease)
- Strong female predominance (F:M = 3-4:1), especially in childbearing years
- Peak onset: 40-60 years
- Black patients: earlier onset, more often diffuse cutaneous form, worse prognosis (more ILD)
- Family history is the strongest risk factor (1.6% of patients have an affected first-degree relative)
- Occupational risk: crystalline silica (sandblasting, mining, ceramics) is the strongest linked exposure; also polyvinyl chloride, aromatic hydrocarbons
The Two Major Subtypes - The Core of SSc
This is the most important classification to know:
| Feature | Limited Cutaneous SSc (lcSSc) | Diffuse Cutaneous SSc (dcSSc) |
|---|---|---|
| Skin distribution | Distal to elbows, face only | Proximal: arms, trunk, face - rapid spread |
| Raynaud | Precedes skin changes by years | Onset coincides with skin changes |
| Visceral onset | Late, mild ILD; PAH as isolated complication (late) | Early, frequent, severe ILD |
| Renal crisis | Very rare | 15%; typically early (<4 years) |
| Calcinosis | Prominent (CREST) | Less common |
| Autoantibody | Anti-centromere (ACA) | Anti-topoisomerase I (Scl-70), Anti-RNA Pol III |
| Prognosis | Better overall | Worse overall |
CREST syndrome is the classic name for lcSSc: Calcinosis, Raynaud, Esophageal dysmotility, Sclerodactyly, Telangiectasia.
Pathogenesis - Three Interlocking Processes
1. Autoimmunity
- CD4+ T cells (Th2-skewed) accumulate in skin and release IL-13 and TGF-β
- IL-13 and TGF-β → fibroblast activation → excess collagen synthesis
- ANAs are present; specific antibodies have diagnostic and prognostic value (see below)
- Type I interferons (IFN signaling genes STAT4, IRF5) play a role
2. Vascular Damage
- Endothelial injury → platelet aggregation → PDGF and TGF-β release → intimal proliferation and perivascular fibrosis
- Progressive obliterative microangiopathy (vasculature narrows and is lost)
- Raynaud phenomenon is the clinical manifestation of vascular dysfunction
- Pulmonary vasculature involvement → pulmonary arterial hypertension (PAH)
3. Fibrosis
- Culmination of macrophage activation, fibrogenic cytokines (TGF-β is the master regulator), and ischemic scarring
- Fibroblasts become constitutively activated, producing excessive Type I and III collagen
Autoantibodies - Must Know
| Antibody | Subtype | Clinical Association |
|---|---|---|
| Anti-centromere (ACA) | lcSSc | CREST features; PAH (late, isolated); better skin prognosis |
| Anti-topoisomerase I (Scl-70) | dcSSc | ILD, diffuse skin involvement; worse prognosis |
| Anti-RNA Polymerase III | dcSSc | Scleroderma renal crisis, rapid skin progression; cancer-associated SSc |
| Anti-U1-RNP | Overlap syndromes | Mixed connective tissue disease |
| Anti-Th/To | lcSSc | PAH, ILD, with limited skin |
ANA is positive in >90% of SSc patients (speckled, nucleolar, or centromere patterns).
Skin Manifestations - Stages
The skin disease evolves through three phases:
Phase 1 - Edematous ("puffy hands"):
- Non-pitting edema of the fingers and hands - the first sign
- Skin is shiny, tight, waxy
- Raynaud phenomenon is typically present
- Patients often complain of "my rings no longer fit"
Phase 2 - Indurative (fibrotic):
- Progressive skin thickening and tightening
- Fibrosis of the dermis, bound to subcutaneous structures
- Hyaline thickening of dermal blood vessel walls
- Thinning of the epidermis, loss of rete pegs, atrophy of adnexal structures
- Loss of normal skin folds; reduced range of motion
Phase 3 - Atrophic:
- Skin becomes atrophic and tightly bound
- Fingers taper and develop "clawlike" appearance (sclerodactyly)
- Face becomes taut and masklike (reduced mouth opening = microstomia)
- Digital ulcers and gangrene from ischemia
- Subcutaneous calcinosis (calcium deposits, especially in CREST)
- Telangiectasia (mat telangiectasias on face, hands, lips)
Organ Involvement
Gastrointestinal (most commonly affected = 90%)
- Esophagus (most severely affected): atrophy of the muscularis → rubber-hose inflexibility of lower 2/3; lower esophageal sphincter dysfunction → GERD, Barrett's esophagus, strictures
- Small bowel: fibrosis → malabsorption, bacterial overgrowth, pseudo-obstruction
- Colon: wide-mouthed diverticula on anti-mesenteric border (characteristic)
Lungs (>50% affected)
- Interstitial lung disease (ILD): NSIP pattern most common (vs UIP in IPF) - insidious dyspnea, bibasal crackles
- Pulmonary arterial hypertension (PAH): especially in lcSSc; progressive dyspnea, cor pulmonale
- ILD + PAH are the leading causes of death in SSc
Kidneys (~30-40%)
- Scleroderma Renal Crisis (SRC): abrupt-onset malignant hypertension + acute renal failure
- Associated with anti-RNA Pol III antibodies and dcSSc (within first 4 years)
- Precipitated by corticosteroids (important!)
- Histology: "onion skin" intimal proliferation of small arteries; fibrinoid necrosis of arterioles
- Treatment: ACE inhibitors (captopril) are life-saving - even in dialysis patients, they can help renal recovery
Heart (1/3 of patients)
- Pericarditis with effusion, myocardial fibrosis, arrhythmias
- Right ventricular failure secondary to pulmonary disease
Musculoskeletal
- Arthralgia and arthritis (early); fibrosis of tendons → tendon friction rubs (leathery crunching on movement) - characteristic of dcSSc
- Inflammatory myositis in ~10%
Raynaud Phenomenon
- Present in >95% of SSc patients
- Classic triphasic: White (vasospasm/ischemia) → Blue (cyanosis/deoxygenation) → Red (reperfusion hyperemia)
- May lead to digital ulcers, pitting scars, and even autoamputation of fingertips
- Distinguish primary Raynaud (benign, common, no autoantibodies) from SSc-associated Raynaud (asymmetric, associated with abnormal nailfold capillaroscopy = dilated/absent capillaries)
Nailfold Capillaroscopy
A simple, non-invasive tool: dilated, distorted, or absent nailfold capillaries (the "scleroderma pattern") strongly suggest CTD-associated Raynaud. Normal nailfold capillaroscopy = primary Raynaud.
Treatment
SSc has no disease-modifying therapy that changes the overall fibrotic course. Management is organ-specific and symptomatic:
| Complication | Treatment |
|---|---|
| Raynaud | CCBs (nifedipine first-line), PDE5 inhibitors (sildenafil), prostacyclin (iloprost) for digital ulcers |
| Digital ulcers | Bosentan (endothelin antagonist) for prevention; phosphodiesterase inhibitors |
| GERD/esophageal | PPIs; prokinetics (metoclopramide); head elevation; avoid large meals |
| ILD | Mycophenolate mofetil (first-line); nintedanib (anti-fibrotic); cyclophosphamide |
| PAH | Endothelin receptor antagonists (bosentan, ambrisentan); PDE5i (sildenafil); prostacyclins |
| Renal crisis | ACE inhibitors (captopril) urgently - this is life-saving |
| Skin/diffuse fibrosis | Methotrexate (limited data); phototherapy for skin |
| Myositis | Corticosteroids (use cautiously - high-dose steroids increase renal crisis risk) |
| Inflammatory arthritis | Low-dose MTX, hydroxychloroquine |
PART 2: DISCOID LUPUS ERYTHEMATOSUS (DLE)
Where DLE Sits in the Lupus Spectrum
The classification of cutaneous lupus is important to understand. DLE is the most common form of chronic cutaneous lupus erythematosus (CCLE):
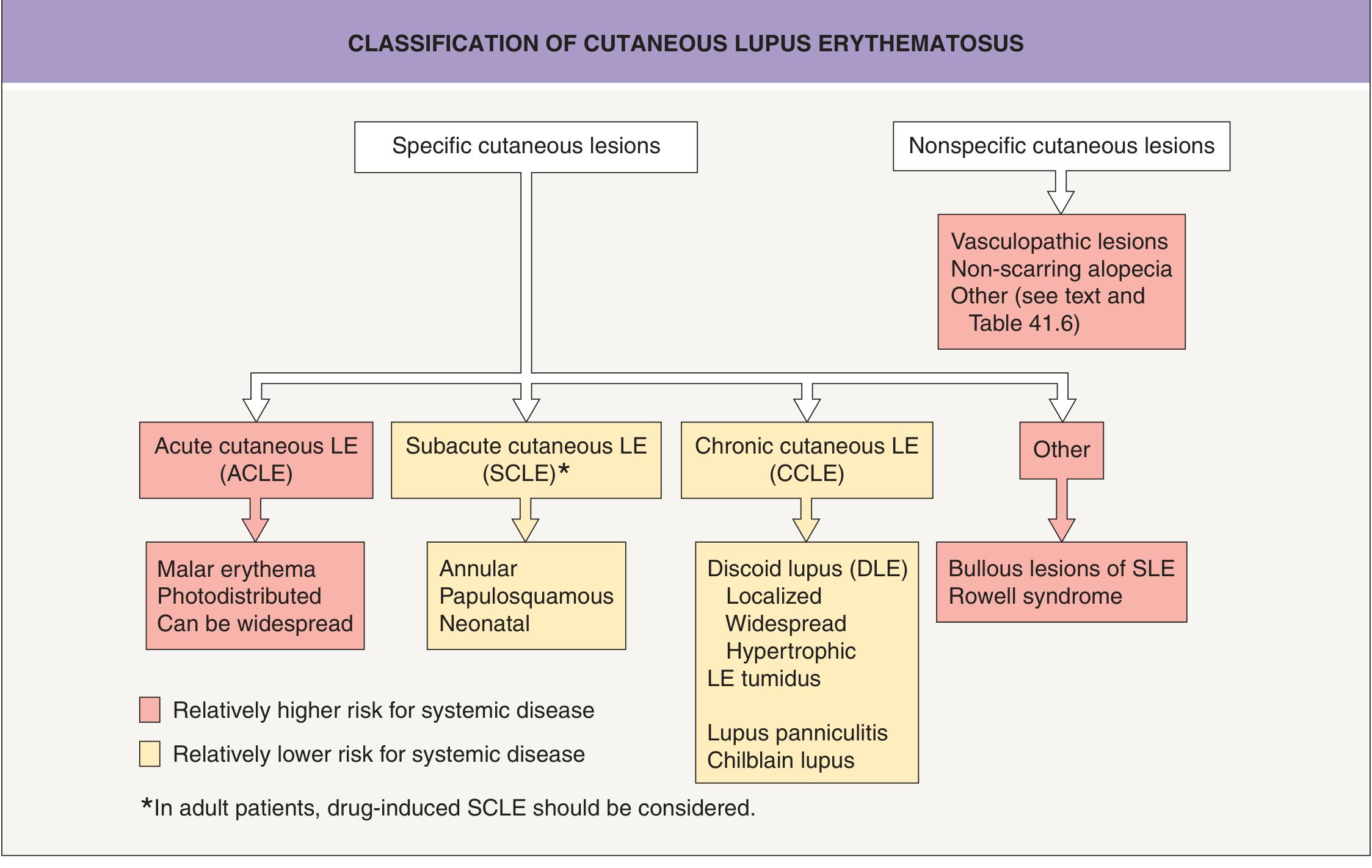
Key classification principle:
- Acute CLE (ACLE): malar rash, photodistributed erythema - highly associated with SLE
- Subacute CLE (SCLE): annular/papulosquamous photodistributed - moderate systemic risk; anti-Ro/SSA positive
- Chronic CLE (CCLE): DLE, lupus panniculitis, tumid lupus - lower systemic risk; primarily a skin disease
Epidemiology
- Most common form of cutaneous lupus
- Young adults; F:M = 2:1
- Can occur in children (higher rate of SLE association in children - up to 26%)
- Localized DLE (above the neck only): only 5-15% progress to SLE
- Generalized DLE (above and below neck): higher risk of SLE, more lab abnormalities
Clinical Features
Classic Presentation: The Three Zones of DLE
Active DLE lesions have a classic tricolor appearance:
- Active erythematous/violaceous border (outer) - active inflammation
- Central atrophy and scarring (middle) - destroyed dermis
- Dyspigmentation (inner) - hypopigmentation centrally, hyperpigmentation peripherally
The evolution: Erythematous macule → indurated plaque with adherent scale → atrophy + scarring + pigment change
Key distinguishing feature: Follicular plugging - hyperkeratosis extends down into patulous follicles, producing "carpet tack" spines on the undersurface of the removed scale (literally looks like carpet tacks). This is pathognomonic.
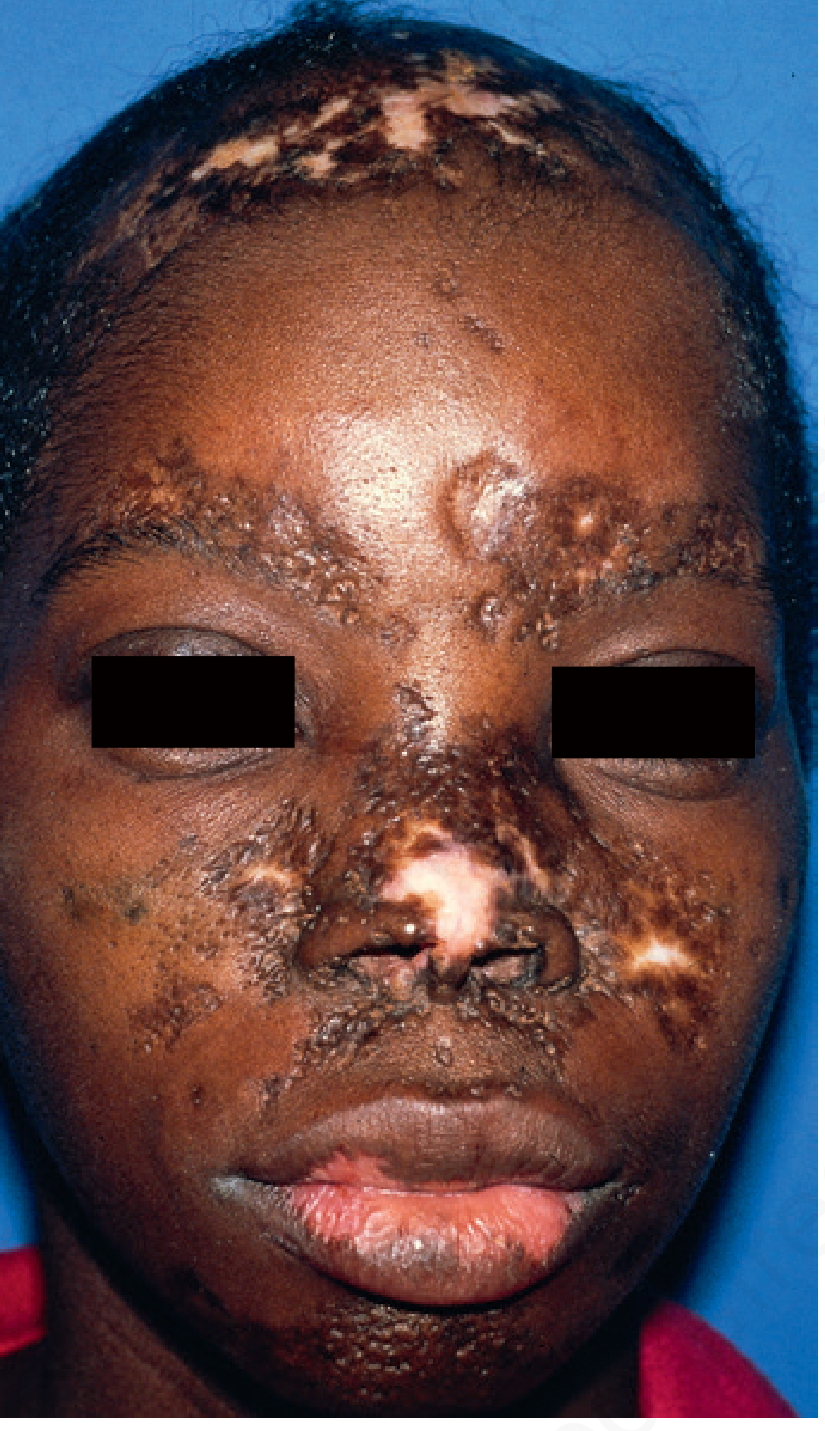
Distribution
- Localized DLE: face (malar areas, nose, ears - particularly the concha of the ear and external canal), scalp, lips
- Lesions on the lips are gray/red and hyperkeratotic with a narrow red inflammatory rim
- Oral/nasal/conjunctival/genital mucosa in ~24% of patients
Scalp DLE - Critical Complication
- Active scalp DLE → permanent scarring alopecia
- Active signs: perifollicular erythema, easily extractable anagen hairs
- End-stage: smooth, depressed white patches or dilated follicular openings in isolated remaining follicles
- This scarring is irreversible - early treatment is paramount
The "Carpet Tack" Sign
When the adherent scale of a DLE lesion is peeled back, the undersurface shows tiny keratotic spines that fit into dilated follicles - like pulling up carpet tacks. This represents follicular plugging and strongly suggests DLE.
Malignant Transformation
Squamous cell carcinoma (SCC) can arise in long-standing, chronic DLE lesions - especially in scars. This is an aggressive SCC with high metastatic potential. Any non-healing lesion within an old DLE scar should raise suspicion.
Histopathology
Changes vary with lesion stage:
Early/acute lesions:
- Vacuolar interface dermatitis (liquefactive degeneration of basal cells)
- Patchy lymphocytic infiltrate
Established lesions (most diagnostic):
- Hyperkeratosis with follicular plugging
- Thickening of the basement membrane (PAS-positive, thickened BMZ - a hallmark)
- Superficial and deep perivascular and periadnexal lymphocytic infiltrate (both above and below - contrast with ACLE/SCLE which are superficial only)
- Dermal mucin deposition
- Vacuolar degeneration of basal cells
- Civatte bodies (like LP, but scattered)
Chronic/inactive lesions:
- Atrophy, fibrosis
- Sparse or absent infiltrate
- Post-inflammatory pigmentation
- Pilosebaceous units destroyed (only "orphaned" arrector pili muscles remain)
Immunofluorescence
- DIF (direct IF) of lesional skin: positive in >75% of established lesions (must be active for several months)
- Pattern: strong, continuous granular deposition of IgG, IgM, and complement (C3) at the DEJ = the "lupus band"
- Contrast with pemphigus (fishnet intercellular) and BP (linear BMZ)
- DIF of non-lesional skin is typically negative in DLE (unlike SLE where non-lesional sun-exposed skin can be positive)
Laboratory Workup
| Test | Expected in Localized DLE | Expected in Generalized DLE / SLE concern |
|---|---|---|
| ANA | Negative or low-titer | Elevated (especially if generalizing) |
| Anti-dsDNA | Negative | Elevated in SLE |
| Anti-Ro/SSA | Negative (positive = SCLE overlap) | Possible |
| ESR | Normal | Elevated |
| CBC | Normal | Leukopenia, anemia |
| Urinalysis | Normal | Proteinuria/hematuria = SLE |
| Complement (C3, C4) | Normal | Low in active SLE |
Differential Diagnosis
DLE must be distinguished from: seborrheic dermatitis, rosacea, psoriasis, sarcoidosis, tinea faciei, actinic keratosis, Bowen disease, lichen planus (esp. on scalp and lips), lupus vulgaris (cutaneous TB), and polymorphic light eruption.
Treatment
Sun protection is the cornerstone - DLE is photosensitive and UV light drives disease activity.
| Therapy | Notes |
|---|---|
| Sun protection | Broad-spectrum SPF 50+ daily; sun avoidance; UPF clothing |
| Topical corticosteroids (first-line) | Superpotent on body; mid-potency on face; intralesional triamcinolone for resistant plaques |
| Hydroxychloroquine (mainstay systemic) | 200-400 mg/day; baseline ophthalmologic exam; requires 2-3 months to see effect; reduces flares and SLE progression |
| Chloroquine | Alternative antimalarial; more retinal toxicity |
| Quinacrine | Added to hydroxychloroquine for resistant disease |
| Topical calcineurin inhibitors | Tacrolimus/pimecrolimus - good for face; steroid-sparing |
| Acitretin or isotretinoin | For hypertrophic or refractory DLE |
| Methotrexate | For widespread or refractory disease |
| Thalidomide | Highly effective for resistant DLE; teratogenicity and neuropathy limit use |
| Dapsone | Bullous or erosive variants |
PART 3: ATOPIC DERMATITIS (ECZEMA)
The Big Concept: A Broken Skin Barrier + Type 2 Inflammation
Atopic dermatitis is not a single disease but a syndrome where a defective epidermal barrier allows allergen sensitization, triggering a Th2-driven immune response that perpetuates inflammation and itch. Understanding the barrier-immune axis is the key to everything in AD.
Epidemiology
- Affects ~20% of children globally; ~10% of adults
- Prevalence still rising in developing nations (plateaued in developed nations in the 1990s)
- Most common chronic skin disease of childhood
- 50% of cases present in the first year of life, almost all within 5 years
- Girls slightly more affected
- Strongly associated with asthma (50%), allergic rhinoconjunctivitis, food allergy = atopic triad
- Atopic march: AD → food allergy/asthma → allergic rhinitis (AD often precedes and may prime this sequence)
Pathogenesis - Two Interlocking Defects
1. Barrier Defect (Filaggrin)
Filaggrin (FLG) gene encodes a protein essential for terminal keratinocyte differentiation and formation of the skin barrier:
- FLG is cleaved by caspase-14 into pyrrolidone carboxylic acid + urocanic acid = "natural moisturizing factor" (NMF)
- FLG null mutations → reduced NMF → xerosis (dry skin)
- Defective lamellar body lipid delivery (especially ceramide deficiency) → impaired lipid bilayers → increased transepidermal water loss (TEWL)
- The leaky barrier allows allergen penetration → sensitization through the skin
- FLG mutations: 42-79% of carriers develop AD; associated with early onset, persistence into childhood, and the atopic march (especially asthma)
- Hyperlinear palms (PPV 71%) and ichthyosis vulgaris are cutaneous markers of FLG mutations
2. Immune Dysregulation (Th2/Th22 inflammation)
- TSLP (thymic stromal lymphopoietin) - produced by keratinocytes - is the "master alarm signal" of the skin that activates dendritic cells and drives Th2 differentiation
- Th2 cells produce IL-4, IL-13 → IgE class-switching, mucus production, inhibit filaggrin expression (perpetuating the barrier defect)
- IL-31 binds directly to cutaneous nerves → itch (key cytokine for pruritus in AD)
- IL-22 (Th22 cells) → epidermal hyperplasia
- JAK-STAT pathway is central to this overactive Th2 signaling
- Secondary: CD8+ T cells and innate lymphoid cells type 2 (ILC2) also contribute
- Staphylococcus aureus colonization (universal in AD) secretes superantigens that massively amplify Th2 inflammation
Why Th2? - This is the opposite of psoriasis (Th17) and LP (Th1). AD is clearly Th2-dominant:
- Elevated total and specific IgE
- Elevated blood eosinophils
- Response to IL-4/IL-13 blockade (dupilumab)
Clinical Features by Age
Stage 1: Infantile AD (birth to 2 years)
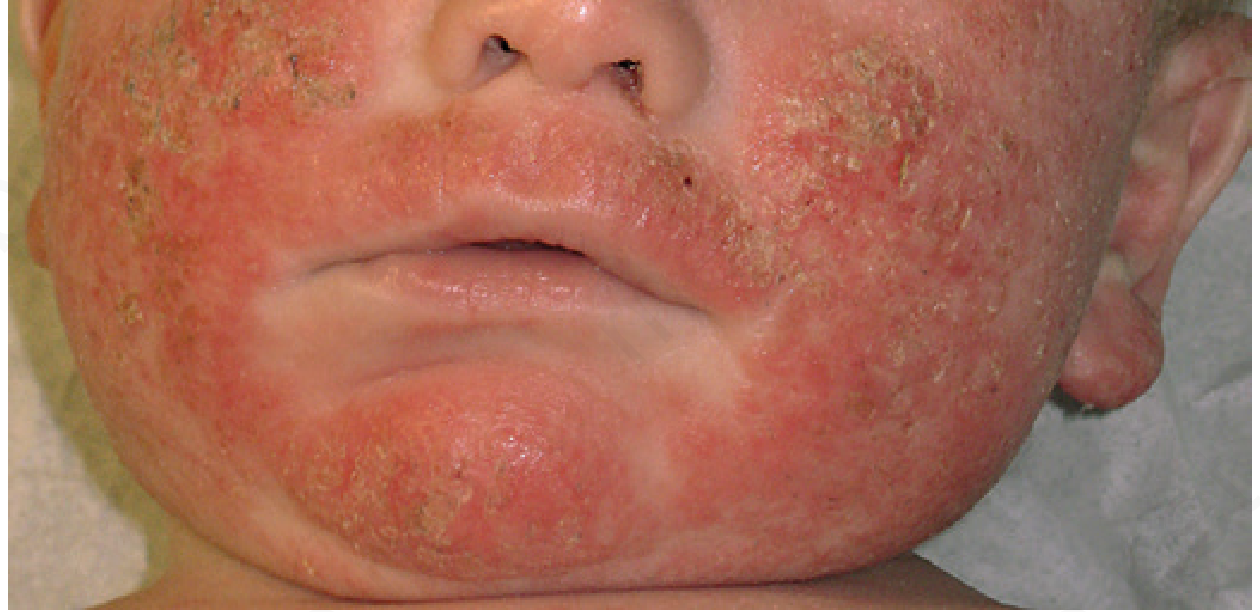
- Begins at 2+ months (never at birth - that would suggest ichthyosis or immunodeficiency)
- Cheeks, forehead, scalp, extensor surfaces - the baby's crawling surfaces
- Weeping, crusting, erythematous patches
- Note: Axillary and inguinal folds typically spared (unlike scabies which involves folds)
- Usually improves by end of year 2
Stage 2: Childhood AD (2-10 years)
- Shifts to flexural surfaces - antecubital and popliteal fossae, flexor wrists, behind knees, ankles
- Less weeping, more lichenified plaques from chronic rubbing
- Nummular morphology common
Stage 3: Adolescent/Adult AD
- Flexural distribution continues; also front of neck, periorbital, perioral
- Lichenification prominent; prurigo-like papules (excoriated firm papules)
- Distribution becomes less characteristic in older adults
- Hand eczema is very common in adults (aggravated by wet work)
- "The itch that rashes" - pruritus often precedes lesions
The "Itch-Scratch Cycle"
This is central to understanding AD:
Itch → Scratching → epidermal barrier disruption → increased allergen/microbe penetration → more inflammation → more itch
Breaking this cycle (with emollients, anti-itch agents, anti-inflammatories) is the therapeutic goal.
Diagnostic Criteria (Hannifin and Rajka)
Major criteria (3 of 4 required):
- Pruritus
- Typical morphology and distribution (flexural in adults; facial/extensor in infants)
- Chronic or chronically relapsing dermatitis
- Personal or family history of atopy (asthma, allergic rhinitis, eczema)
Minor criteria (supporting, many listed): xerosis, ichthyosis, elevated IgE, early onset, keratosis pilaris, Dennie-Morgan fold, Hertoghe sign, white dermatographism, pityriasis alba, nipple eczema, food hypersensitivity, etc.
Physical Signs of the Atopic Diathesis
| Sign | Description |
|---|---|
| Dennie-Morgan fold | Transverse infraorbital skin fold; indicative of atopic constitution |
| Hertoghe sign | Thinning/loss of lateral third of eyebrows from chronic rubbing |
| White dermatographism | Scratching produces white line (not red as in normal skin) - paradoxical vasoconstriction |
| "Headlight sign" | Extensive facial eczema sparing the nose (characteristic of AD) |
| Keratosis pilaris (KP) | Rough follicular papules on upper arms, thighs, cheeks - FLG mutation marker |
| Hyperlinear palms | Accentuated palmar creases from FLG mutations |
| Pityriasis alba | Hypopigmented, mildly scaly patches on cheeks in children (subclinical dermatitis) |
Complications
- Eczema herpeticum (Kaposi varicelliform eruption): HSV dissemination through the broken barrier → widespread punched-out erosions, fever, potentially fatal. Contraindication to smallpox vaccination (vaccinia = severe, potentially fatal).
- Staph aureus superinfection: universal colonization; exacerbates AD through superantigen production; treat with mupirocin topically or systemic antibiotics for overt infection
- Molluscum contagiosum: widespread, exuberant in atopics
- Eczema vaccinatum: disseminated vaccinia from exposure to smallpox vaccine
Treatment - Stepwise Approach
Step 1: Skin Care Foundation (All Patients)
- Emollients/moisturizers: applied liberally and frequently, within 3 minutes of bathing ("soak and smear"); ointments preferred (no preservatives, better occlusion)
- Bathing: lukewarm soaks 10-15 min to hydrate skin; gentle non-soap cleansers
- Avoid triggers: wool, harsh soaps, sweat, known allergens, stress
- Reduce Staph colonization: dilute bleach baths (0.005% sodium hypochlorite - "bleach baths")
Step 2: Anti-inflammatory Therapy
Topical corticosteroids (TCS) - cornerstone:
- Ointments preferred over creams
- Low-potency for face, genitals, skin folds (hydrocortisone 1-2.5%)
- Mid-potency for body (triamcinolone 0.1%)
- High-potency for thick, lichenified plaques (clobetasol) - short courses only
- Once daily application nearly as effective as twice daily; do not over-apply
- Steroid phobia is common - address parental concerns directly
- Maintenance: 2-5x weekly application to frequently-flaring areas
Topical calcineurin inhibitors (TCIs) - steroid-sparing:
- Tacrolimus 0.03% / 0.1% ointment (Protopic)
- Pimecrolimus 1% cream (Elidel)
- No skin atrophy (unlike steroids) - preferred for face, eyelids, skin folds
- Safe for long-term use; burning on application common initially
- Not for age <2 years (tacrolimus 0.03%); not for immunocompromised
Topical PDE4 inhibitor:
- Crisaborole - non-steroid, non-TCI option; mild-moderate AD
Step 3: Systemic Therapy (Moderate-Severe)
| Agent | Mechanism | Notes |
|---|---|---|
| Dupilumab (Dupixent) | Anti-IL-4Rα (blocks IL-4 + IL-13) | Paradigm-shifting biologic; safe long-term; SC q2 weeks; FDA approved 6 months+; also treats asthma and nasal polyps |
| Tralokinumab | Anti-IL-13 | Approved moderate-severe AD adults |
| JAK inhibitors (upadacitinib, abrocitinib) | JAK1 inhibition → suppress Th2 | Oral; rapid onset; black box warnings (thrombosis, malignancy) |
| Cyclosporine | Calcineurin inhibitor (systemic) | Rapid, effective; limited to 1-2 years (nephrotoxicity) |
| Methotrexate | Anti-inflammatory | Slower onset; second-line systemic |
| Azathioprine | Immunosuppressive | Long-term use; TPMT testing before starting |
| Mycophenolate mofetil | Anti-proliferative | Alternative to cyclosporine/MTX |
Phototherapy: NB-UVB (first-line phototherapy); UVA1 (especially for lichenified/fibrotic disease)
Dupilumab - The Key Biologic
Dupilumab targets IL-4Rα (the shared receptor subunit for IL-4 and IL-13), blocking both cytokines simultaneously. Since IL-4 and IL-13 are the master regulators of the Th2 response in AD, this is highly effective. Response rates: EASI-75 (75% reduction in eczema severity) in ~50-60% of patients. Also approved for asthma and CRS with nasal polyps.
THREE-WAY COMPARISON TABLE
| Feature | Systemic Sclerosis | DLE | Atopic Dermatitis |
|---|---|---|---|
| Type | Autoimmune CTD | Chronic cutaneous lupus | Allergic/atopic skin disease |
| Key mechanism | Th2 + TGF-β fibrosis; vasculopathy | Interface dermatitis (T-cell vs basal cells) | Barrier defect (FLG) + Th2 inflammation |
| Primary lesion | Skin induration/thickening | Indurated disc plaque with follicular plugging | Eczematous patches/lichenification |
| Scarring | Progressive fibrosis/atrophy | YES - permanent (key danger) | No scarring |
| Mucosal | GI primarily; oral telangiectasia | Oral in ~24% | No |
| Autoantibodies | Anti-centromere, Scl-70, RNA Pol III | ANA (low titer), lupus band on DIF | No specific autoantibodies; elevated IgE |
| Key organs | Lung (ILD/PAH), kidney, GI | Skin only (mostly) | Skin; associations with asthma, rhinitis |
| Hallmark histology | Dermal fibrosis, obliterative vasculopathy | BMZ thickening, follicular plugging, deep perivascular infiltrate | Spongiosis, perivascular eosinophils, hyperkeratosis |
| Prognosis | Variable; ILD/PAH/SRC = fatal | Good if treated early; SCC risk in old scars | Chronic relapsing; most improve with age |
| Cornerstone Rx | Organ-specific (ACE-I for SRC; MMF for ILD) | Sun protection + hydroxychloroquine | Emollients + TCS + dupilumab for severe |
Memory Tricks
- SSc = "Skin becomes Stone"** - fibrosis turns skin into hard, tight stone; remember CREST for limited form
- SSc autoantibodies: lcSSc = "Centromere in CREST"; dcSSc = "Scl-70 in dcSSc" (both start with hard 'S' sounds)
- DLE: Discs, Dyspigmentation, Death of follicles (scarring alopecia), Direct IF positive at DEJ
- AD = "Inside Out": broken barrier lets things IN (allergens), and water escapes OUT (TEWL); Th2 is the T-helper that says "Be Allergic" - type 2 = atopy
- Atopic march: A-A-A-R = AD → Asthma → Allergic rhinitis (in that order)
- Dupilumab = Du-pi = blocks IL-4 and IL-13 (the two Th2 cytokines)
Sources: Robbins & Kumar Basic Pathology, Harrison's Principles of Internal Medicine 22e, Goldman-Cecil Medicine, Dermatology 2-Volume Set 5e, Andrews' Diseases of the Skin
This is a shared conversation. Sign in to Orris to start your own chat.