Raas system
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Finding Sources
Reading File
Reading File
Reading File
Loading Image
I now have everything needed for a thorough answer. Here is the complete overview of the RAAS:
Renin-Angiotensin-Aldosterone System (RAAS)
The RAAS is a multi-organ hormonal cascade that regulates blood pressure, extracellular fluid (ECF) volume, sodium/potassium balance, and renal hemodynamics. It is one of the most powerful long-term blood pressure control systems in the body.
Cascade Flowchart
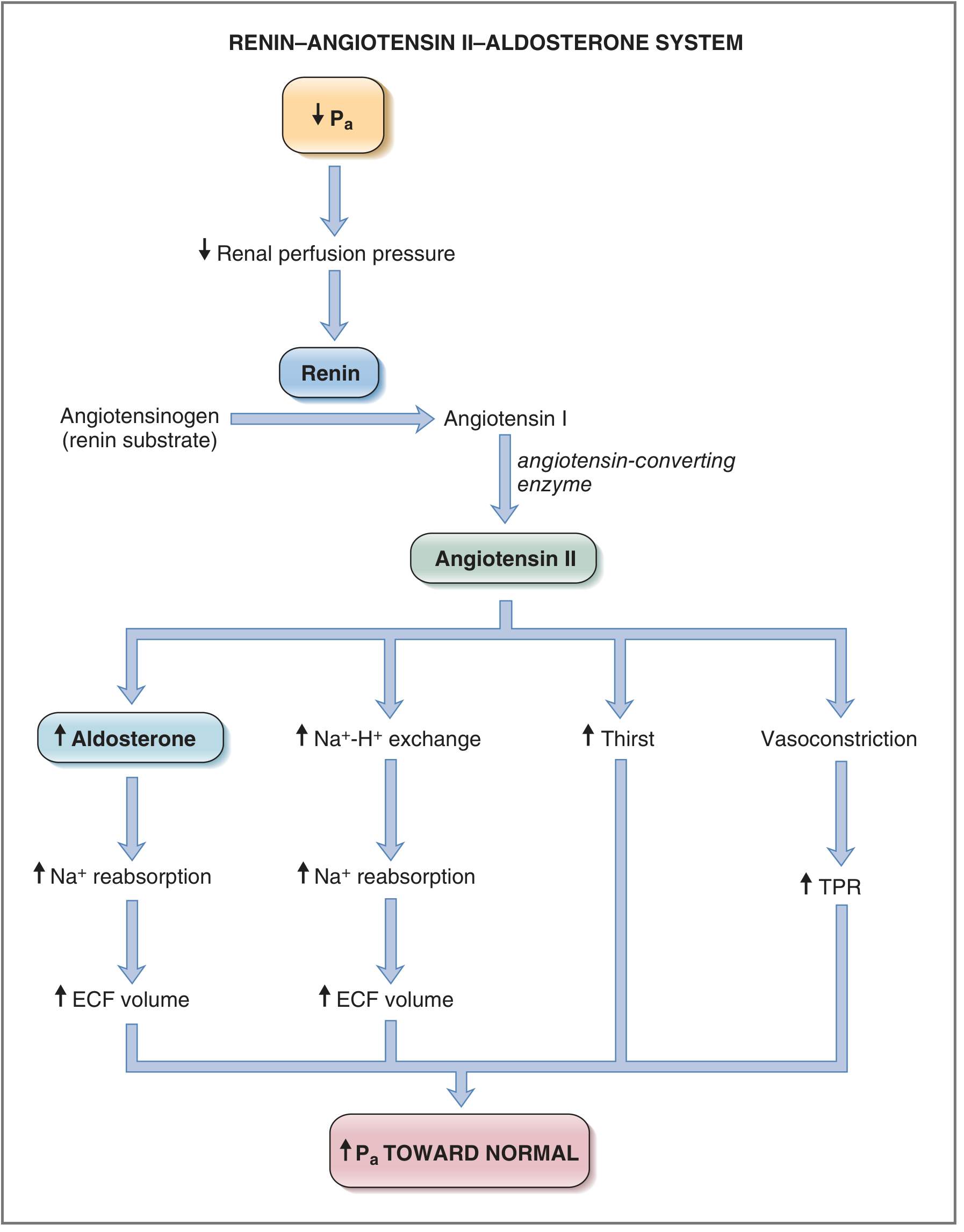
Step-by-Step Mechanism
1. Renin Release (Kidney - Juxtaglomerular Apparatus)
The juxtaglomerular (JG) cells of the renal afferent arterioles release renin - an aspartyl protease - in response to:
- Decreased renal perfusion pressure (e.g., hypotension, hemorrhage, dehydration)
- Low blood sodium (sensed by the macula densa)
- Sympathetic nervous system stimulation (via beta-1 adrenergic receptors)
- Upright posture
2. Angiotensinogen → Angiotensin I (Liver + Blood)
Renin cleaves angiotensinogen (an alpha-2 globulin produced by the liver) to form the inactive decapeptide angiotensin I.
3. Angiotensin I → Angiotensin II (Lungs)
Angiotensin-converting enzyme (ACE), located primarily in pulmonary endothelium, converts angiotensin I into the active octapeptide angiotensin II - one of the most potent vasoconstrictors in the human body.
Actions of Angiotensin II
Angiotensin II acts through AT1 receptors (Gq protein-coupled, via IP3/Ca²+ second messenger) to produce four major effects:
| Target | Effect | Result |
|---|---|---|
| Adrenal cortex (zona glomerulosa) | Stimulates aldosterone synthesis (via cholesterol desmolase + aldosterone synthase) | Na⁺ retention, K⁺ excretion, ECF volume expansion |
| Renal proximal tubule | Stimulates Na⁺-H⁺ exchange | Na⁺ and HCO₃⁻ reabsorption |
| Hypothalamus | Increases thirst + stimulates ADH secretion | Increased water intake and reabsorption |
| Arterioles | Vasoconstriction | Increased total peripheral resistance (TPR) |
Role of Aldosterone
Aldosterone is secreted by the zona glomerulosa of the adrenal cortex. It acts on the principal cells of the distal tubule and collecting duct to:
- Increase Na⁺ reabsorption
- Increase K⁺ excretion
- Retain water (ECF volume expansion)
Because aldosterone requires gene transcription and new protein synthesis, its effects take hours to days - making the RAAS a slower compensatory system compared to the rapid baroreceptor reflex.
Primary controls of aldosterone secretion:
- Angiotensin II (main driver) - via renin-angiotensin axis
- Serum K⁺ - hyperkalemia directly depolarizes adrenal cells and opens voltage-gated Ca²+ channels, stimulating aldosterone
- ACTH - provides a tonic permissive effect (stimulates the first step, cholesterol desmolase)
Net Outcome (Feedback Loop)
A decrease in mean arterial pressure (Pa) → ↑ Renin → ↑ Angiotensin II → ↑ Aldosterone:
- Na⁺ retention → ECF expansion → ↑ blood volume → ↑ cardiac output → ↑ Pa
- Arteriolar vasoconstriction → ↑ TPR → ↑ Pa
- ↑ ADH + thirst → ↑ water reabsorption → further ECF expansion
As Pa restores, renal perfusion pressure rises and renin release is suppressed - completing the negative feedback loop.
Pharmacological Targets (RAAS Inhibitors)
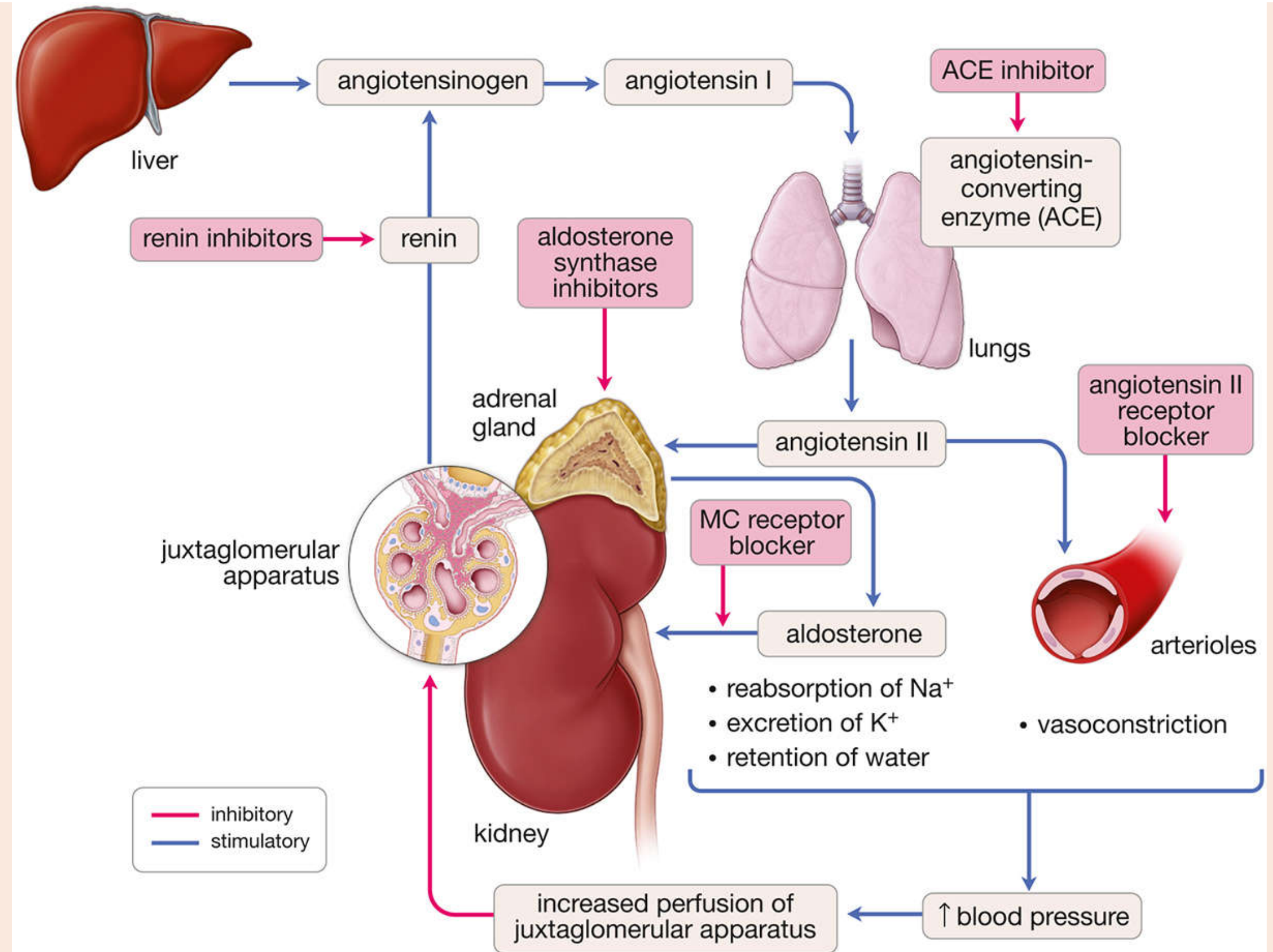
| Drug Class | Example Drugs | Site of Action |
|---|---|---|
| ACE inhibitors | Captopril, Enalapril, Lisinopril | Block conversion of Ang I → Ang II in the lungs |
| Angiotensin Receptor Blockers (ARBs) | Losartan, Valsartan | Block AT1 receptors on target tissues |
| Renin inhibitors | Aliskiren | Block renin directly, preventing Ang I generation |
| Mineralocorticoid receptor blockers | Spironolactone, Eplerenone | Block aldosterone receptor in the kidney |
| Aldosterone synthase inhibitors | (Experimental) | Block final step of aldosterone biosynthesis |
Clinical note: ACE inhibitors were developed after a factor in the venom of the Brazilian pit viper (Bothrops jararaca) was found to potently inhibit ACE. The "lesion" in chronic essential hypertension is now believed to be excessive angiotensin II production in the lung. - Histology: A Text and Atlas, p. 1889
Clinical Relevance
- Hypertension: Excessive RAAS activity sustains elevated blood pressure; RAAS blockade is a cornerstone of antihypertensive therapy
- Heart failure: Angiotensin II and aldosterone worsen cardiac remodeling; ACE inhibitors/ARBs reduce mortality
- Diabetic nephropathy: RAAS blockade (ACE inhibitors or ARBs) slows progression independent of blood pressure effects - Harrison's Principles, 22e
- Salt-sensitive hypertension: Impaired RAAS suppression during high salt intake drives blood pressure sensitivity - Guyton & Hall Medical Physiology
- Primary hyperaldosteronism (Conn's syndrome): Autonomous aldosterone excess causes hypertension + hypokalemia
Sources: Costanzo Physiology 7e | Histology: A Text and Atlas | Guyton & Hall Textbook of Medical Physiology | Harrison's Principles of Internal Medicine 22e | Brenner & Rector's The Kidney
What happens to RAAS in kidney disease or secondary hypertension?
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Finding Sources
Reading File
Reading File
I have comprehensive content. Now I'll compose the full answer.
RAAS in Kidney Disease and Secondary Hypertension
The RAAS sits at the center of both causing and perpetuating kidney disease and hypertension. The relationship runs in both directions: kidney disease activates the RAAS, and chronic RAAS overactivation damages the kidneys further - a vicious cycle.
1. Renovascular Hypertension - The Goldblatt Model
The clearest example of RAAS-driven secondary hypertension is renal artery stenosis (RAS). The mechanism was first proven in Goldblatt's classic experiments: constricting a renal artery in dogs caused hypertension proportional to the degree of narrowing.
Causes of Renal Artery Stenosis
- Atherosclerosis (70% of cases) - typically occurs at the origin of the renal artery; more common in older males and diabetics
- Fibromuscular dysplasia (FMD) - fibrous/fibromuscular thickening of the arterial wall; more common in young women (3rd-4th decade); shows a "string of beads" appearance on angiography
The Two-Kidney, One-Clip Model (Unilateral RAS)
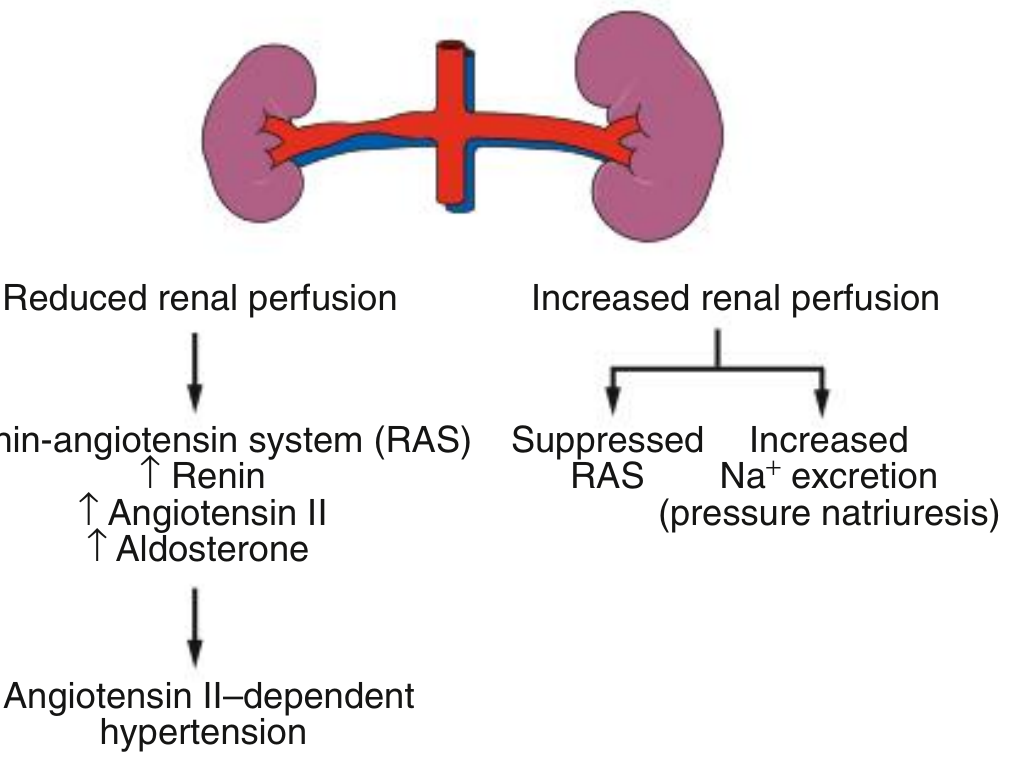
| Side | What Happens |
|---|---|
| Stenotic kidney | Reduced perfusion pressure → ↑ renin → ↑ Angiotensin II → ↑ Aldosterone |
| Contralateral kidney | Receives increased perfusion pressure → suppressed RAS → pressure natriuresis (Na⁺ excretion) |
The result is angiotensin II-dependent hypertension - maintained primarily by renin and angiotensin, not volume. Plasma renin activity is elevated, and BP falls dramatically with ACE inhibitors or ARBs. However, in this setting, ACE inhibition can cause the GFR of the stenotic kidney to fall (because it is angiotensin II-dependent on efferent arteriolar tone to maintain glomerular filtration). - Comprehensive Clinical Nephrology, 7e
Bilateral RAS or Solitary Kidney with RAS (One-Kidney, One-Clip)
When both kidneys are ischemic (or there is only one kidney with stenosis), there is no normal kidney to excrete sodium. The pathophysiology shifts:
- Renin and angiotensin levels do not remain persistently elevated
- Hypertension becomes sodium and volume dependent rather than angiotensin-dependent
- Other pressor pathways take over: sympatho-adrenergic activation, oxidative stress, and impaired vasodilatory responses - Campbell Walsh Wein Urology, 3-Volume Set
2. RAAS in Chronic Kidney Disease (CKD)
CKD creates a self-reinforcing cycle driven by RAAS overactivation:
Nephron loss → Reduced renal perfusion → ↑ Renin → ↑ Angiotensin II
↓ ↓
Glomerular hypertension ←── Efferent arteriolar constriction ←──┘
↓
Proteinuria → Inflammation → TGF-β ↑ → Fibrosis → Further nephron loss
The key mechanisms are:
Hemodynamic injury: Angiotensin II selectively constricts the efferent arteriole, raising intraglomerular capillary pressure. This increases the hydrostatic pressure across the glomerular basement membrane, causing proteinuria and glomerular damage.
Fibrotic injury: RAAS activation upregulates TGF-β (transforming growth factor-beta), which drives mesangial matrix accumulation, inflammation, and interstitial fibrosis. This is especially prominent in diabetic nephropathy, where hyperglycemia amplifies this process via protein kinase C (PKC).
Systemic hypertension: Angiotensin II and aldosterone cause sodium retention and vasoconstriction, sustaining systemic hypertension which adds to glomerular injury.
This is why RAAS blockade is renoprotective on three levels simultaneously: hemodynamic (reducing glomerular hypertension), antifibrotic (blunting TGF-β), and antiproteinuric. - National Kidney Foundation Primer on Kidney Diseases, 8e
Therapies for Slowing CKD Progression (Proven Benefit)
| Therapy | Mechanism |
|---|---|
| ACE inhibitors | Block Ang II production; dilate efferent arteriole; reduce proteinuria |
| ARBs | Block AT1 receptor; same hemodynamic benefit |
| Selective MR antagonists (spironolactone, eplerenone) | Block aldosterone's fibrotic/inflammatory effects |
| SGLT2 inhibitors | Reduce glomerular hyperfiltration; independent RAAS-modulatory effects |
| BP control (target 120-140/80-90 mmHg) | Reduce transmission of systemic pressure to glomeruli |
3. Primary Aldosteronism (Conn Syndrome) - RAAS Bypassed at the Bottom
This is an important cause of secondary hypertension where the problem is autonomous aldosterone excess, not driven by renin or angiotensin:
- Cause: Adrenal adenoma (zona glomerulosa) or bilateral adrenal hyperplasia secreting excess aldosterone autonomously
- Effect: Na⁺ retention → ECF and blood volume expansion → hypertension + hypokalemia + mild metabolic alkalosis
Key diagnostic hallmark: LOW plasma renin
Because volume expansion suppresses the JG cells, renin is suppressed by negative feedback. This is the opposite of renovascular hypertension. The aldosterone-to-renin ratio (ARR) is elevated.
Other features:
- Hypokalemia (can cause muscle weakness or paralysis)
- Plasma Na⁺ mildly elevated (usually <4-6 mEq/L increase)
- Occasional polydipsia/hyponatremia (dipsogenic effect of Ang II is absent, yet volume drives thirst via other mechanisms)
Treatment: Surgical removal of adenoma, or spironolactone/eplerenone for hyperplasia. - Guyton & Hall Medical Physiology
4. Full Classification of Secondary Hypertension Causes
| Category | Examples | RAAS Role |
|---|---|---|
| Renal parenchymal | Glomerulonephritis, diabetic nephropathy, polycystic kidney disease, hydronephrosis | RAAS overactivated due to reduced perfusion |
| Renovascular | Renal artery stenosis (atherosclerotic or FMD), aortic coarctation, Takayasu arteritis | Renin-angiotensin excess (2K1C) or volume-dependent (bilateral) |
| Renoprival | End-stage kidney disease, anephric state | Loss of pressure natriuresis; volume-dependent |
| Endocrine | Primary aldosteronism (low renin), renin-secreting tumor (high renin), Cushing syndrome, pheochromocytoma, acromegaly | Varies - primary aldosteronism has suppressed RAAS; renin-secreting tumor has overdriven RAAS |
| Drugs | NSAIDs (block prostaglandins that normally promote renin), cyclosporine, glucocorticoids, oral contraceptives | Various mechanisms |
Secondary hypertension accounts for <10% of all hypertension cases. - Fuster & Hurst's The Heart, 15e
Summary: RAAS States in Different Conditions
| Condition | Renin | Angiotensin II | Aldosterone | BP |
|---|---|---|---|---|
| Unilateral RAS | ↑↑ | ↑↑ | ↑↑ | ↑↑ |
| Bilateral RAS (chronic) | Normal/↓ | Normal/↓ | Normal/↓ | ↑↑ (volume-driven) |
| Primary aldosteronism | ↓↓ | ↓ | ↑↑ | ↑↑ |
| CKD (early-mid) | ↑ | ↑ | ↑ | ↑ |
| Heart failure (with RAAS activation) | ↑ | ↑ | ↑ | Low/normal (but RAAS harmful) |
| Renin-secreting tumor | ↑↑↑ | ↑↑↑ | ↑↑↑ | ↑↑↑ |
Sources: Comprehensive Clinical Nephrology 7e | Robbins, Cotran & Kumar Pathologic Basis of Disease | National Kidney Foundation Primer on Kidney Diseases 8e | Guyton & Hall Medical Physiology | Fuster & Hurst's The Heart 15e | Campbell Walsh Wein Urology
This is a shared conversation. Sign in to Orris to start your own chat.