atherosclerosis in biochemistry
Reading File
Finding Sources
Searching PubMed
atherosclerosis biochemistry pathogenesis
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
I now have comprehensive content from the biochemistry textbooks. Let me compile the full detailed answer.
Atherosclerosis - Biochemistry
Atherosclerosis is a chronic, progressive disease of arterial walls driven by lipid accumulation, oxidative stress, and inflammatory signaling. From a biochemistry standpoint, it is best understood through its molecular initiators, the lipid chemistry involved, the role of modified lipoproteins, and the cellular cascade that follows.
1. Definition and the Arterial Wall
The arterial wall has three layers: the tunica intima (innermost, lined by endothelium), tunica media (smooth muscle), and adventitia (outermost connective tissue). Atherosclerosis develops primarily in the intima.
2. Risk Factors - Biochemical Basis
The following biochemical/metabolic derangements can injure the vascular endothelium, initiating the process:
- Elevated LDL cholesterol, VLDL remnants, and chylomicron remnants in the blood
- Low HDL (reverse cholesterol transport is impaired)
- Elevated blood glucose (chronic hyperglycemia promotes peroxynitrite production, which drives lipid oxidation)
- Cigarette smoking (generates reactive oxygen species)
- Elevated angiotensin II (vasoconstricting and pro-inflammatory)
- Hypertension (mechanical stress on endothelium)
Basic Medical Biochemistry - A Clinical Approach 6e, p. 1211
3. Central Biochemical Mechanism: LDL Oxidation
The single most important biochemical event is the oxidative modification of LDL particles.
-
LDL particles entering the subendothelial space are exposed to reactive oxygen species (superoxide, nitric oxide, hydrogen peroxide) generated by vascular cells.
-
These ROS oxidize the polyunsaturated fatty acids (PUFAs) within LDL via lipid peroxidation - a free radical chain reaction:
- Initiation: A free radical (X•) abstracts a hydrogen from a PUFA → fatty acid radical (R•)
- Propagation: R• + O₂ → peroxyl radical (ROO•) → attacks another PUFA → forms lipid peroxide + new radical (chain reaction, potentially peroxidizing thousands of PUFA molecules)
- Termination: Two radicals combine → non-radical product
-
The result is oxidized LDL (oxLDL) - a structurally and functionally altered lipoprotein that drives atherogenesis.
Harper's Illustrated Biochemistry 32e, p. 225
4. Step-by-Step Pathogenesis (Biochemical Cascade)
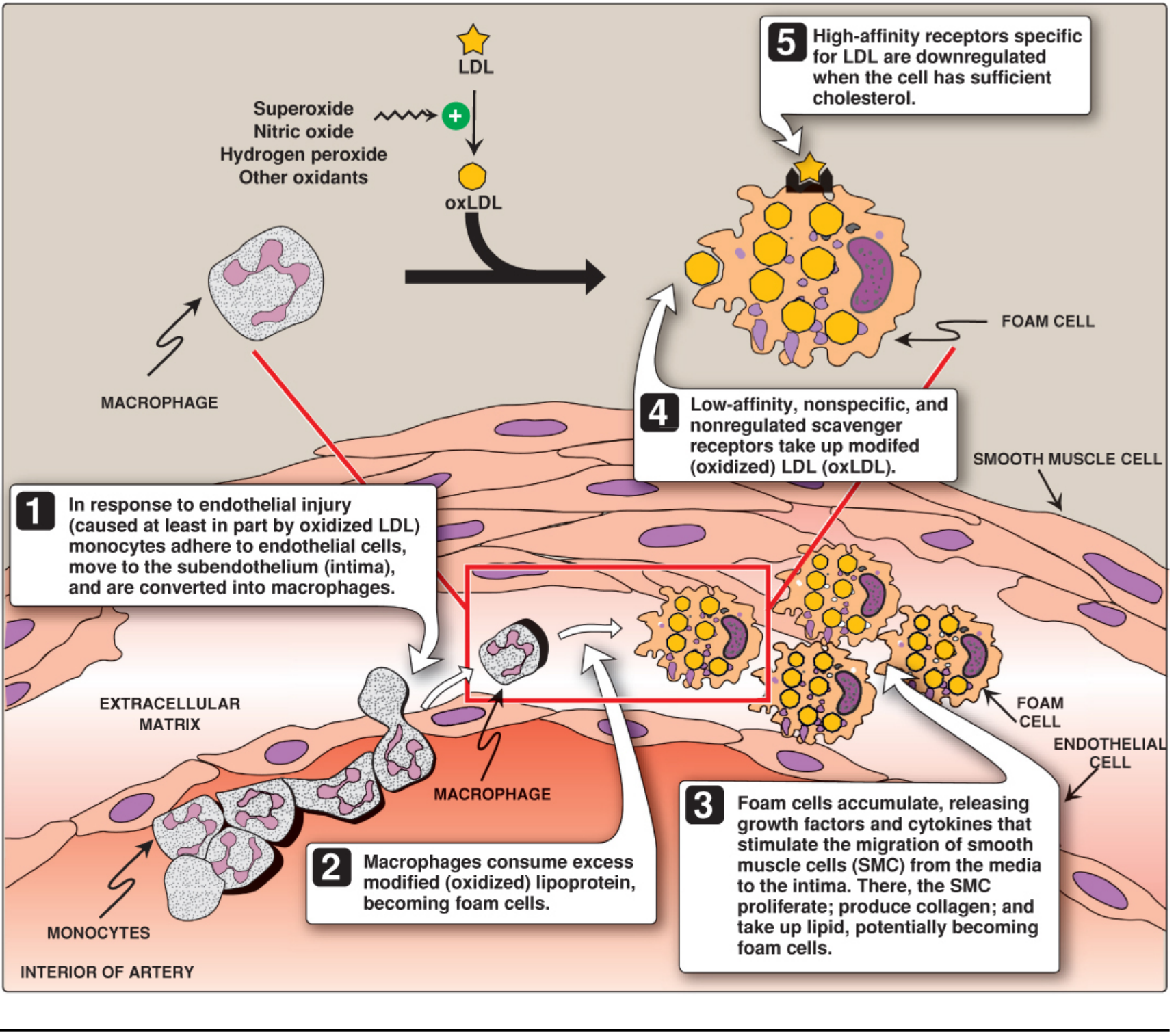
Step 1 - Endothelial Injury & Monocyte Adhesion
OxLDL injures endothelial cells, triggering them to express adhesion molecules: VCAM-1 (vascular cell adhesion molecule-1) and ICAM-1 (intercellular adhesion molecule-1). These capture circulating monocytes, slowing their movement and allowing them to enter the intima through spaces between endothelial cells.
Step 2 - Macrophage Transformation (Foam Cell Formation)
Monocytes in the intima differentiate into macrophages. These macrophages express scavenger receptors (SR-A type) - low-affinity, non-specific, unregulated receptors that recognize and avidly take up oxLDL. (This contrasts with classical LDL receptors on liver cells, which are tightly regulated by cellular cholesterol content and are downregulated when cholesterol is sufficient.) Because scavenger receptors are not downregulated, macrophages continue taking up oxLDL indefinitely, becoming lipid-engorged foam cells.
Step 3 - Fatty Streak Formation
Accumulating foam cells form the fatty streak - the earliest visible lesion. These appear as yellow-white linear streaks that bulge slightly into the lumen. Foam cells release growth factors (e.g., PDGF) and cytokines that:
- Stimulate migration of smooth muscle cells (SMCs) from the tunica media into the intima
- SMCs proliferate, produce collagen, and also take up lipid - potentially becoming foam cells themselves
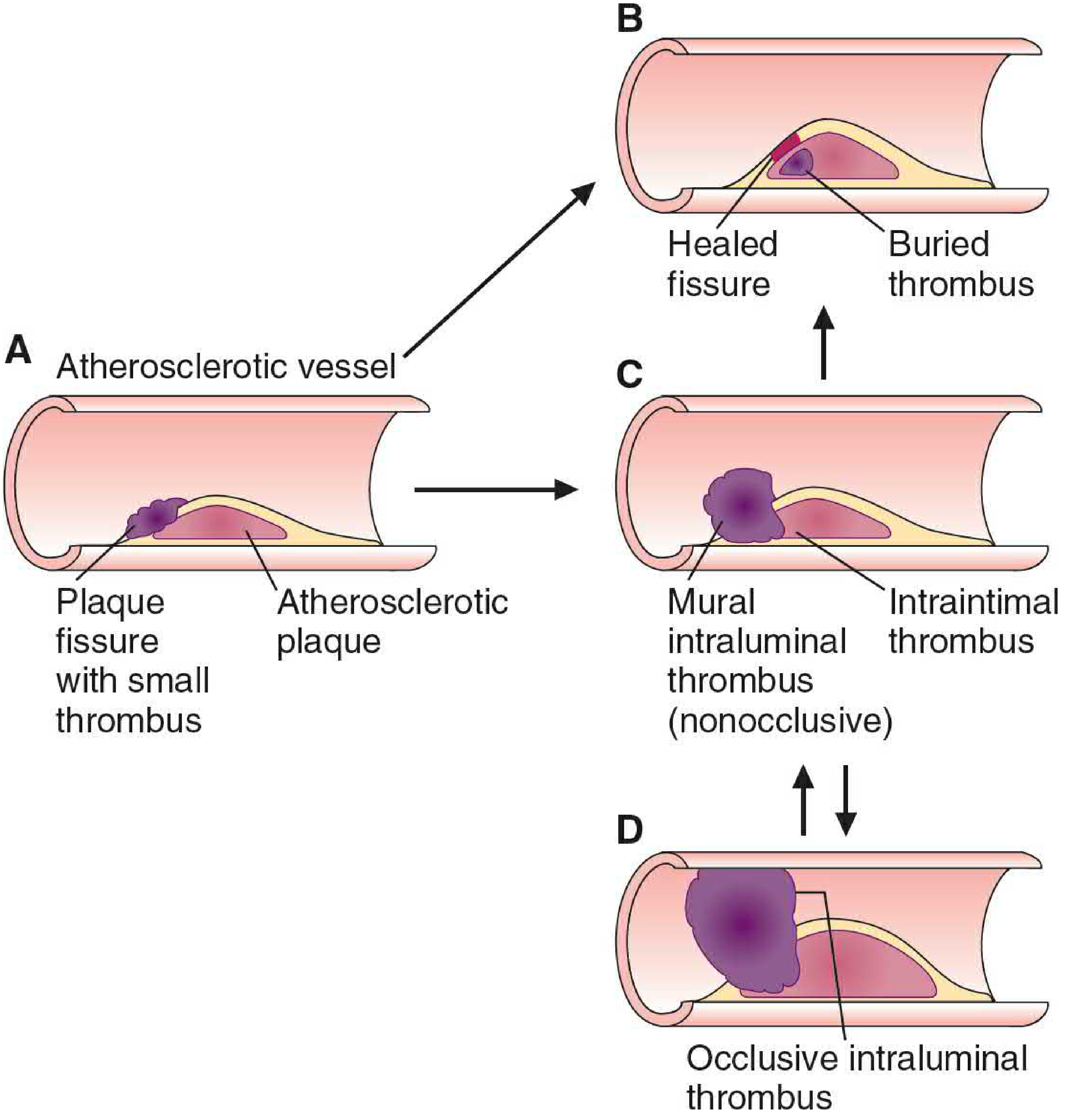
Step 4 - Fibrous Plaque Development
As the plaque matures, a fibrous cap forms - rich in collagen secreted by SMCs and macrophages. The necrotic lipid core beneath the cap contains cholesterol crystals, dead foam cells, and calcium deposits (driven by oxidized sterols and TGF-β inducing osteogenic gene expression).
Step 5 - Plaque Rupture and Thrombosis
SMCs and macrophages secrete matrix metalloproteinases (MMPs) that degrade collagen in the fibrous cap, particularly at the shoulders ("elbows") of the plaque. When the thinned cap ruptures, the procoagulant plaque contents contact circulating blood, triggering platelet activation and acute thrombus formation. If the thrombus fully occludes the lumen, myocardial infarction or stroke results.
Basic Medical Biochemistry 6e, pp. 1211-1213
5. The LDL Receptor - Key Biochemical Regulator
The LDL receptor (6 structural domains) is responsible for normal LDL clearance from blood via receptor-mediated endocytosis through clathrin-coated pits:
| Domain | Structure | Function |
|---|---|---|
| 1st | Cysteine-rich repeats | Binds apo B-100 on LDL |
| 3rd | N-linked oligosaccharides | Glycosylation |
| 4th | O-linked sugars (Ser/Thr-rich) | Extends receptor from membrane |
| 5th | 22 hydrophobic residues | Membrane-spanning anchor |
| 6th | Cytosolic C-terminal | Interacts with clathrin-coated pit |
Regulation: When cellular cholesterol is adequate, LDL receptor synthesis decreases (downregulation). This feedback loop is broken by the scavenger receptor pathway, which macrophages use to take up oxLDL without limit.
Familial Hypercholesterolemia (FH): Mutations in the LDL receptor gene impair this uptake:
- Heterozygotes (~1/300): ~50% of normal receptors, elevated LDL, premature atherosclerosis
- Homozygotes (~1/million): nearly no LDL receptors, serum LDL >500 mg/dL, severe early atherosclerosis
Four classes of mutations exist: null alleles (no protein), transport defects (protein not reaching cell surface), binding defects, and internalization defects.
Basic Medical Biochemistry 6e, pp. 1209-1211
6. HDL and Reverse Cholesterol Transport (Protective Mechanism)
HDL is "good cholesterol" because it performs reverse cholesterol transport (RCT):
- Nascent (disc-shaped) HDL, synthesized by liver and intestine, accepts free cholesterol from peripheral cells (including arterial wall cells) via the ABCA1 transporter
- Cholesterol is immediately esterified by LCAT (lecithin:cholesterol acyltransferase), activated by apo A-I, maintaining the concentration gradient for continued efflux
- Cholesteryl ester-rich HDL2 returns to liver, where SR-B1 (scavenger receptor B type 1) mediates selective cholesteryl ester uptake
- CETP (cholesteryl ester transfer protein) transfers some cholesteryl esters from HDL to VLDL in exchange for triglyceride
Low HDL is an independent risk factor for atherosclerosis. HDL levels are raised by exercise, estrogen, and moderate alcohol; low HDL often accompanies elevated VLDL/LDL.
Lippincott's Biochemistry 8e, p. 658; Basic Medical Biochemistry 6e, p. 1212
7. Lipoprotein(a) [Lp(a)] - Additional Biochemical Risk Factor
Lp(a) is an LDL-like particle with an additional apolipoprotein, apo(a), covalently linked to apo B-100. Apo(a) is structurally homologous to plasminogen (the fibrin-dissolving protease precursor) - suggesting Lp(a) may competitively inhibit fibrinolysis, promoting thrombosis on top of plaques. Lp(a) levels are primarily genetically determined; niacin is one of the few agents that reduces Lp(a).
Lippincott's Biochemistry 8e, p. 660
8. Inflammation as the Unifying Theme
Atherosclerosis is recognized as a chronic inflammatory disorder at its core:
- Monocyte accumulation in the intima resembles classical inflammation
- Foam cells secrete pro-inflammatory cytokines (TNF-α, IL-1, IL-6) and growth factors
- Aspirin (inhibits COX-mediated prostaglandin synthesis, reduces platelet activation) and statins (HMG-CoA reductase inhibitors - reduce cholesterol and suppress the inflammatory cascade) both work partly through anti-inflammatory mechanisms
9. Pharmacological Interventions - Biochemical Basis
| Drug | Biochemical Target | Effect |
|---|---|---|
| Statins (e.g., atorvastatin) | Inhibit HMG-CoA reductase | ↓ cholesterol synthesis → ↑ LDL receptor expression → ↓ blood LDL |
| Ezetimibe | Blocks NPC1L1 in intestinal brush border | ↓ cholesterol absorption from intestine |
| Niacin | Inhibits VLDL secretion from liver | ↓ LDL, ↓ VLDL, ↓ Lp(a); ↑ HDL |
| Aspirin | Inhibits COX → ↓ TXA₂ | Reduces platelet aggregation at plaque rupture sites |
Summary Diagram of Biochemical Pathogenesis
Elevated LDL / Hypertension / Smoking / Hyperglycemia
↓
Endothelial Injury
↓
LDL enters subintima → Oxidized by ROS (superoxide, H₂O₂, NO) → oxLDL
↓
Endothelial cells express VCAM-1, ICAM-1
↓
Monocyte adhesion → Transmigration → Differentiate into Macrophages
↓
Scavenger receptors take up oxLDL (unregulated) → Foam Cells
↓
Fatty Streak → Fibrous Plaque (SMC migration, collagen deposition)
↓
MMP secretion → Fibrous cap thinning → Plaque Rupture
↓
Platelet activation → Thrombus → MI / Stroke
Sources:
- Basic Medical Biochemistry - A Clinical Approach, 6e (Marks et al.), pp. 1209-1213
- Biochemistry, 8th ed - Lippincott Illustrated Reviews, pp. 655-660
- Harper's Illustrated Biochemistry, 32e, pp. 225-226
- Robbins & Kumar Basic Pathology, Pathogenesis of Atherosclerosis
This is a shared conversation. Sign in to Orris to start your own chat.