Teach me barter syndrome in Paediatrics
Reading File
Finding Sources
Searching PubMed
Bartter syndrome children diagnosis management
Reading File
Reading File
Reading File
Reading File
Loading Image
Finding Sources
Reading File
Reading File
Finding Sources
I now have comprehensive information to write a detailed, well-organized teaching response on Bartter syndrome in paediatrics.
Bartter Syndrome in Paediatrics
Overview
Bartter syndrome (BS) is a rare group of autosomal recessive renal tubular disorders characterized by impaired salt reabsorption in the thick ascending limb (TAL) of the loop of Henle. The hallmark biochemical features are:
- Hypokalemia
- Hypochloremic metabolic alkalosis
- Hyperreninemia and hyperaldosteronism
- Normal-to-low blood pressure (vascular hyporesponsiveness to angiotensin II)
- Elevated urinary prostaglandin E2
It was first described by Dr. Frederic Bartter in patients who presented in childhood, which is why the postnatal form is termed "classic" Bartter syndrome.
Pathophysiology
The core defect is loss-of-function in one of several transporters responsible for NaCl absorption in the TAL. This mimics the effect of a loop diuretic (like furosemide/bumetanide).
The TAL - Normal Function
In the TAL, sodium is reabsorbed via the apical NKCC2 cotransporter (Na⁺-K⁺-2Cl⁻). This requires:
- Potassium recycling back into the lumen via apical ROMK channels - which maintains a lumen-positive voltage that drives paracellular reabsorption of Na⁺, Ca²⁺, and Mg²⁺
- Exit of Cl⁻ on the basolateral side via ClC-Ka and ClC-Kb chloride channels, supported by the accessory subunit Barttin
What Happens When These Fail
Loss of TAL salt reabsorption causes:
- Salt (NaCl) wasting → volume depletion
- Activation of the renin-angiotensin-aldosterone system (RAAS)
- Increased NaCl delivery to the distal nephron and collecting duct → enhanced K⁺ and H⁺ secretion → hypokalemia and metabolic alkalosis
- Impaired chloride reabsorption at the macula densa → activation of COX-2 → elevated prostaglandin E2 → juxtaglomerular apparatus hypertrophy → further hyperreninism + afferent arteriolar dilation
- Despite high aldosterone, blood pressure is LOW due to vascular hyporesponsiveness to angiotensin II
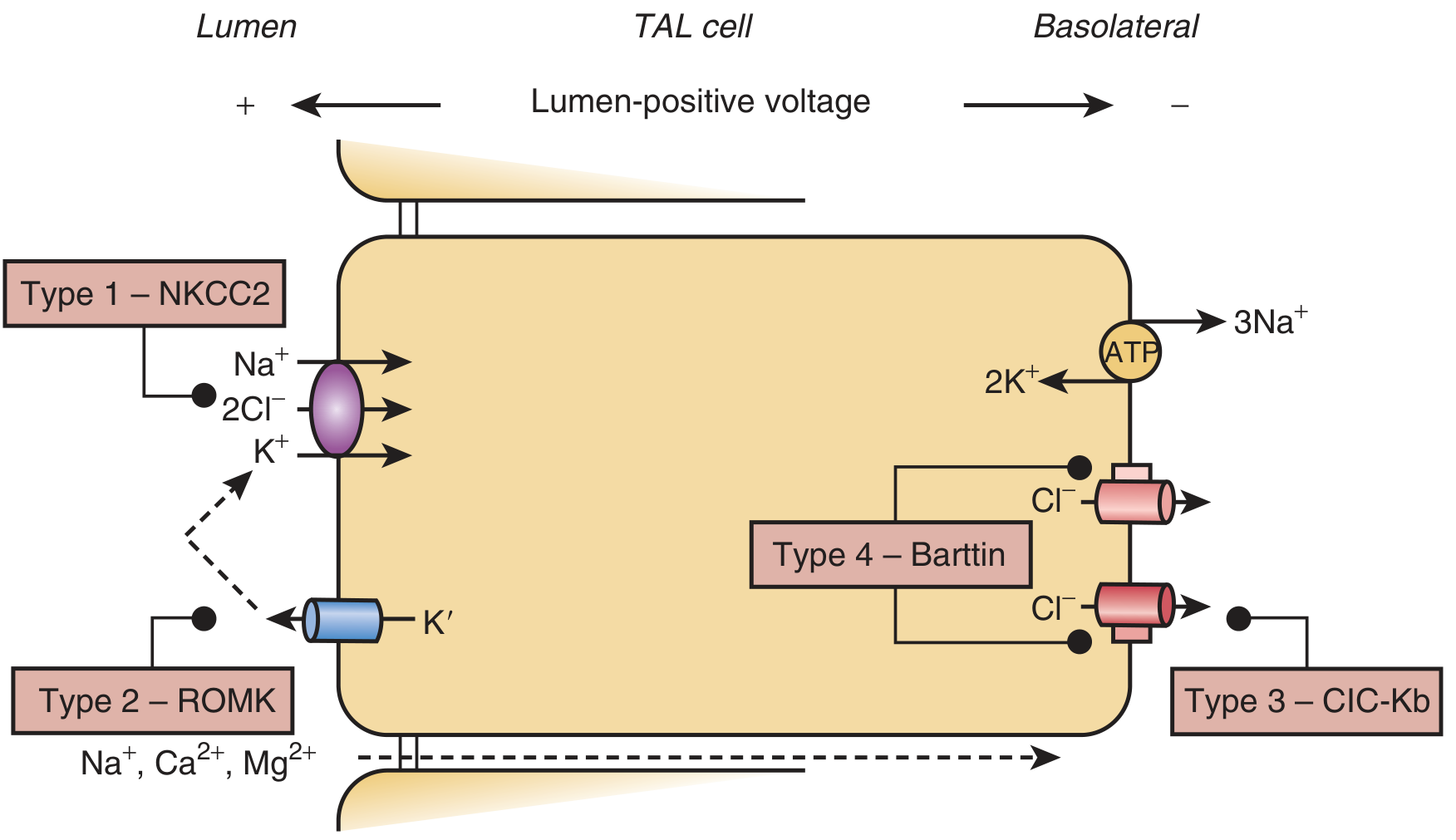
Schematic of a TAL cell showing the four transporter defects. Type 1 = NKCC2 (apical); Type 2 = ROMK (apical K⁺ channel); Type 3 = ClC-Kb (basolateral Cl⁻ channel); Type 4 = Barttin subunit (basolateral). - Brenner and Rector's The Kidney
Genetic Classification
| Type | Gene | Protein | Mutation Effect | Typical Presentation |
|---|---|---|---|---|
| Type 1 | SLC12A1 (NKCC2) | Na⁺-K⁺-2Cl⁻ cotransporter | Loss of primary cotransport | Antenatal/neonatal |
| Type 2 | KCNJ1 (ROMK) | Renal outer medullary K⁺ channel | Loss of apical K⁺ recycling | Antenatal/neonatal |
| Type 3 | CLCNKB (CLC-Kb) | Basolateral Cl⁻ channel | Loss of basolateral Cl⁻ exit | Classic/postnatal (milder) |
| Type 4a | BSND (Barttin) | Barttin accessory subunit | CLC-Ka and CLC-Kb both fail | Antenatal + sensorineural deafness |
| Type 4b | CLCNKA + CLCNKB | Both Cl⁻ channels | Combined channel loss | Severe antenatal |
| Type 5 | CASR (gain-of-function) | Calcium-sensing receptor | Inhibits ROMK and NKCC2 in TAL | Variable |
| Transient | MAGED2 | MAGED2 protein | Reduced expression of NKCC2 and NCC in utero | Transient antenatal, resolves at 3-4 weeks |
Key point: Type 3 (CLCNKB) is the classic childhood presentation. Types 1, 2, and 4 are more severe and typically present antenatally or in the neonatal period.
Clinical Presentations
Antenatal / Neonatal Form (Types 1, 2, 4)
- Polyhydramnios in utero (fetal polyuria)
- Premature birth
- Postnatal polyuria and polydipsia (nephrogenic diabetes insipidus-like picture)
- Vomiting, failure to thrive
- Hypercalciuria → nephrocalcinosis (calcium deposited in the kidneys)
- Profound electrolyte disturbances (hypokalemia, metabolic alkalosis)
- Type 4 specifically: associated with sensorineural deafness (Barttin is also expressed in K⁺-secreting cells of the inner ear)
Special notes:
- Type 2 (ROMK) may paradoxically present with hyperkalemia in the neonatal period (ROMK also mediates K⁺ secretion in the collecting duct - its loss transiently impairs K⁺ excretion). This resolves in the first week as other K⁺ channels mature. Can be mistakenly diagnosed as pseudohypoaldosteronism type 1.
- MAGED2 mutations cause a severe but transient antenatal form that resolves completely by 3-4 weeks of life.
Classic Form (Type 3, also Types 1/2 with milder mutations)
- Presents in childhood (or even adulthood for mild mutations)
- Polyuria, polydipsia
- Failure to thrive
- Muscle weakness, fatigue, cramps
- Hypokalemic metabolic alkalosis
- Hyperreninemia, hyperaldosteronism
- Normal-to-low blood pressure (never hypertensive)
- Hypercalciuria (but LESS than antenatal form; nephrocalcinosis is rare in type 3)
- About 20% are hypomagnesemic
Key Biochemistry
| Parameter | Result |
|---|---|
| Serum K⁺ | LOW (hypokalemia) |
| Serum Cl⁻ | LOW |
| Arterial pH / bicarbonate | HIGH (metabolic alkalosis) |
| Blood pressure | Normal-to-low |
| Plasma renin | HIGH |
| Plasma aldosterone | HIGH |
| Urine K⁺ | HIGH (renal K⁺ wasting) |
| Urine Cl⁻ | HIGH (not low like in vomiting) |
| Urine Ca²⁺ | HIGH (hypercalciuria) - esp. types 1, 2, 4 |
| Serum Mg²⁺ | Normal (unlike Gitelman) |
| Urinary prostaglandin E2 | HIGH |
Diagnosis
Approach
- Clinical suspicion - child with hypokalemia, metabolic alkalosis, polyuria/polydipsia, normal or low BP, failure to thrive
- Biochemistry - confirm hypokalemia, metabolic alkalosis, elevated renin and aldosterone, hypercalciuria, elevated urine Cl⁻
- Exclude other causes:
- Vomiting/laxative abuse: urine Cl⁻ is LOW in vomiting (Cl⁻ is retained), but HIGH in Bartter
- Diuretic abuse: history + diuretic screen
- Loop diuretics mimic the exact biochemical picture of Bartter syndrome
- Genetic testing - confirms subtype; not always required in clinical practice but increasingly used
Distinguishing Bartter from Gitelman Syndrome
| Feature | Bartter | Gitelman |
|---|---|---|
| Defect location | TAL (loop of Henle) | DCT (distal convoluted tubule) |
| Analogous to | Loop diuretic effect | Thiazide diuretic effect |
| Onset | Neonatal/childhood | Adolescence/adulthood |
| Severity | More severe | Milder |
| Urine calcium | HIGH (hypercalciuria) | LOW (hypocalciuria) |
| Serum magnesium | Usually normal | LOW (hypomagnesemia) |
| Nephrocalcinosis | Common (types 1, 2, 4) | Absent |
| Polyuria/polydipsia | Prominent | Absent |
| Urinary PGE2 | Elevated | Normal |
| Mutated gene | NKCC2, ROMK, CLCNKB, BSND | SLC12A3 (NCC) |
The distinguishing rule: Bartter = hypercalciuria + normal Mg²⁺. Gitelman = hypocalciuria + hypomagnesemia.
Complications
- Nephrocalcinosis - in antenatal/neonatal types due to severe hypercalciuria
- Growth retardation / failure to thrive (partly due to chronic electrolyte imbalance and high prostaglandins)
- Cardiac arrhythmia - from severe hypokalemia
- Muscle paralysis - from profound hypokalemia
- Growth hormone deficiency, hypophosphatemia, hyperparathyroidism (associated in some)
- Sensorineural deafness (type 4)
- Nephrogenic diabetes insipidus - some type 1/2 patients develop profound hyposthenuria; salt supplementation helps but can worsen water losses in NDI
Treatment
There is no cure. Treatment aims to replace electrolyte losses and reduce the pathological consequences of RAAS activation and excess prostaglandin production.
1. Salt and Potassium Replacement
- Oral NaCl and KCl supplementation - the mainstay in all types
- Target serum K⁺ > 3 mEq/L
- In acute antenatal/neonatal crisis: IV saline infusions for prolonged periods to avoid dehydration
2. COX Inhibitors (NSAIDs)
- Indomethacin 2-4 mg/kg/day in 2-4 divided doses - most commonly used, especially for antenatal/perinatal types (1, 2, 4, 5)
- Reduces COX-2-driven prostaglandin overproduction → ameliorates polyuria, electrolyte wasting, and improves growth
- Also increases plasma K⁺ and decreases plasma renin activity
- Does NOT correct the underlying tubular defect
- Adverse effects: bowel perforation, necrotizing enterocolitis (especially in neonates)
- Selective COX-2 inhibitors (celecoxib, rofecoxib) have been used as alternatives; improvement in growth and phosphate wasting reported with celecoxib. Concern about cardiovascular risk in adults has dampened enthusiasm, though relevance to pediatric BS patients is unclear.
- Urinary prostaglandin E2 levels guide response to treatment
3. Potassium-Sparing Diuretics
- Spironolactone - blocks aldosterone; reduces kaliuresis
- Amiloride - blocks ENaC in collecting duct
- Use with extreme caution in infants - these drugs worsen salt wasting and risk life-threatening hypovolemia. Best reserved for severe cases with arrhythmia or paralysis from hypokalemia.
4. Magnesium
- Supplement if hypomagnesemic (more common in type 3 and Gitelman)
5. ACE Inhibitors / ARBs
- Captopril has been used to counter the high angiotensin II activity - adjunct option in some refractory cases
Summary Mnemonic: "BARTTER"
- Blood pressure - normal or low (never hypertensive)
- Alkalosis - metabolic, hypochloremic
- Renin - elevated (secondary hyperreninism)
- Thick ascending limb - site of the defect
- Transporter mutations - NKCC2, ROMK, ClC-Kb, Barttin
- Electrolyte wasting - K⁺ (and Na⁺, Cl⁻, Ca²⁺)
- Recessive - autosomal recessive inheritance
Sources
- Brenner and Rector's The Kidney, Chapters 44 and 73
- National Kidney Foundation Primer on Kidney Diseases, 8e, Chapter 14
- Goldman-Cecil Medicine, International Edition
- Recent review: Fang C & Zhou W, Pediatr Nephrol 2025 (PMID 39283520) - Genetic background of neonatal hypokalemia
This is a shared conversation. Sign in to Orris to start your own chat.