Carcinogenesis
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
I now have extensive material from Robbins Pathologic Basis of Disease and Robbins Basic Pathology to construct a thorough answer. Let me compile the full response.
Carcinogenesis
Carcinogenesis is the process by which normal cells undergo malignant transformation - acquiring the capacity for uncontrolled proliferation, tissue invasion, and metastasis. It is fundamentally a genetic disease driven by the stepwise accumulation of mutations and epigenetic alterations.
1. Fundamental Principles
Nonlethal genetic damage is the foundation of carcinogenesis. Initial damage may arise from:
- Environmental exposures (chemicals, radiation, viruses)
- Inherited germline mutations
- Spontaneous/random mutations ("bad luck")
Key features of carcinogenesis:
- Clonal origin: a tumor arises from a single precursor cell that acquired genetic damage; all daughter cells carry the same mutations
- Multistep process: no single mutation is fully transforming; multiple complementary driver mutations must accumulate over time
- Driver mutations: mutations that directly contribute to the cancer hallmarks; the first is called the initiating mutation, maintained in all cells; subsequent "hits" are required for full malignancy
- Robbins, Cotran & Kumar Pathologic Basis of Disease
2. Four Target Gene Classes
Four classes of genes are the principal targets of cancer-causing mutations:
| Gene Class | Effect of Mutation | Genetic Behavior |
|---|---|---|
| Proto-oncogenes | Gain-of-function → excessive growth promotion | Dominant (one mutant allele sufficient) |
| Tumor suppressor genes | Loss-of-function → remove growth brakes | Recessive (both alleles usually needed) |
| Apoptosis-regulating genes | Loss-of-function → enhanced cell survival | Variable |
| DNA repair genes | Loss-of-function → accelerated mutation rate ("mutator phenotype") | Recessive; indirect effect |
3. Oncogenes and Growth Promotion
Oncoproteins are constitutively active versions of normal growth-signaling molecules. They arise through:
- Point mutations (e.g., RAS mutations - found in ~20-30% of all cancers)
- Gene amplification (e.g., NMYC amplification in neuroblastoma, appearing as homogenous staining regions or double minutes)
- Chromosomal translocations (e.g., BCR-ABL in CML)
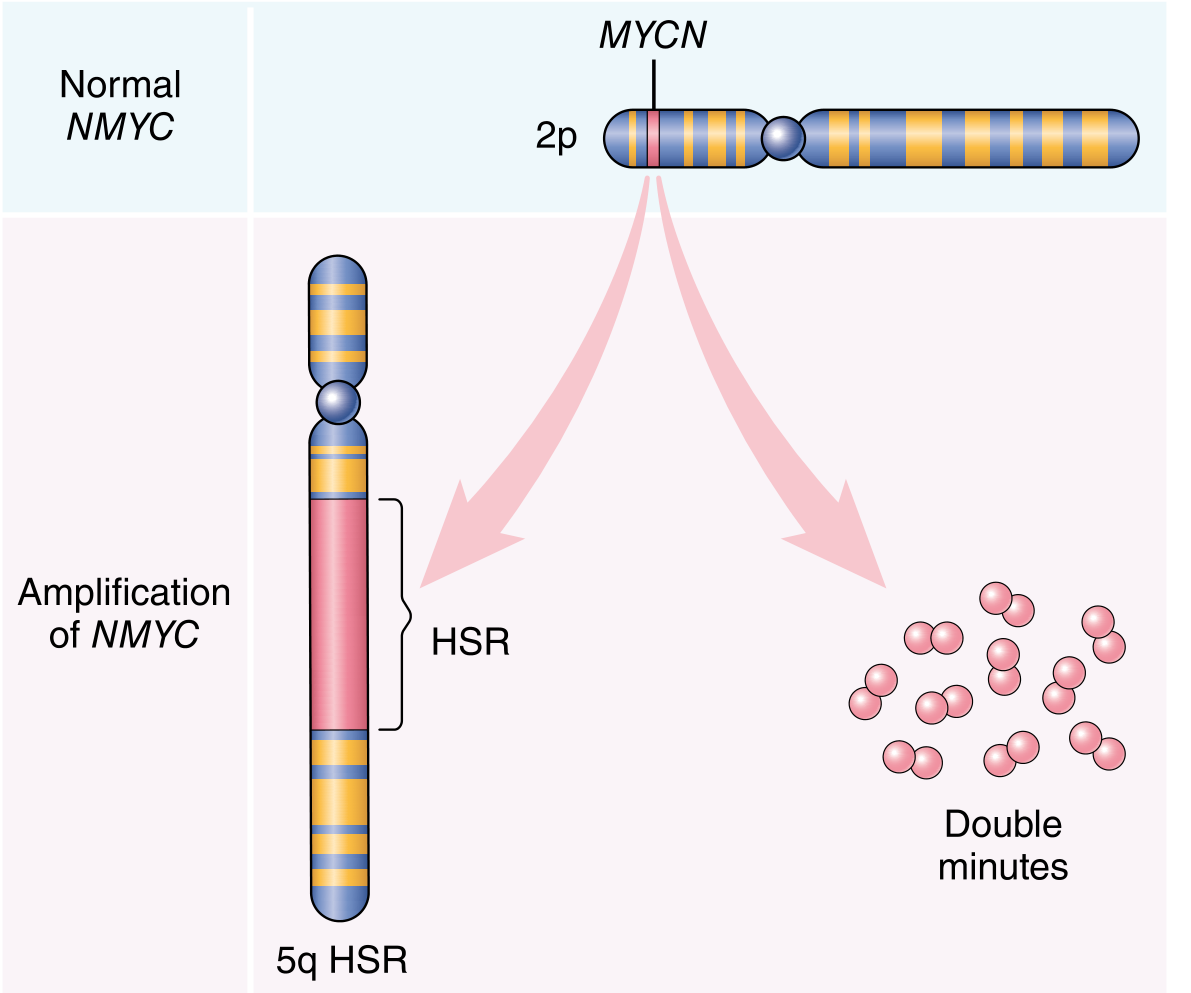
Key oncoproteins by category:
| Category | Example | Mechanism | Associated Cancer |
|---|---|---|---|
| Growth factors | TGF-α | Overexpression (autocrine loop) | Astrocytomas |
| Growth factor receptors | ERBB1 (EGFR) | Activating mutation | Lung adenocarcinoma |
| Growth factor receptors | ERBB2 (HER2) | Amplification | Breast carcinoma |
| Signal transducers | RAS | Point mutation | Many carcinomas |
| Transcription factors | MYC | Translocation/amplification | Burkitt lymphoma, neuroblastoma |
| Cell cycle proteins | Cyclin D, CDK4 | Amplification | Many cancers |
RAS acts as a key signaling node: growth factor receptors activate RAS, which normally cycles between active (GTP-bound) and inactive (GDP-bound) states. Mutant RAS is locked in the GTP-bound (active) state, continuously driving proliferation via MAP kinase and PI3K/AKT pathways.
4. Tumor Suppressor Genes
RB: "Governor of the Cell Cycle"
- Normally, hypophosphorylated RB binds and sequesters E2F transcription factors, blocking G1→S transition
- Growth signals → cyclin D/CDK4 → RB hyperphosphorylation → E2F release → cell cycle progression
- RB is inactivated in cancers by:
- Loss-of-function RB mutations (retinoblastoma: "two-hit hypothesis")
- Amplification of cyclin D or CDK4
- Loss-of-function mutations in CDK inhibitors (e.g., p16/INK4a)
- Viral oncoproteins (e.g., HPV E7, polyomavirus large T antigen) that bind and inactivate RB
TP53: "Guardian of the Genome"
- The most frequently mutated gene in human cancers
- Activated by DNA damage, hypoxia, and oncogene activation
- p53 induces:
- p21 (CDK inhibitor) → cell cycle arrest
- BAX → apoptosis
- Senescence programs
- Anti-angiogenic molecules (thrombospondin-1)
- Loss of p53 removes all these safeguards and promotes angiogenesis
Other Key Tumor Suppressors
- PTEN: Brake on PI3K/AKT signaling; mutated in Cowden syndrome and many sporadic cancers
- VHL: Targets HIF-1α for degradation under normoxia. Loss → HIF-1α accumulates → VEGF/PDGF upregulation → angiogenesis and Warburg metabolism. Mutated in hereditary and sporadic renal cell carcinoma
- TGF-β pathway: Normally activates CDK inhibitors and represses MYC; pathway lost in colorectal, stomach, endometrial, and pancreatic cancers
- APC/β-catenin: Lost early in colorectal carcinogenesis
Cell cycle component mutations in cancer (summarized):
| Component | Role | Cancer Relevance |
|---|---|---|
| CDK4/Cyclin D | Phosphorylate RB; allow G1/S transition | Amplified in many cancers |
| p16/INK4a (CDKN2A) | Blocks CDK4, preserves RB function | Deleted/silenced in many cancers |
| p21 (CDKN1A) | Induced by p53; arrests cell cycle | Lost in p53-mutant cancers |
| p27 (CDKN1B) | Responds to TGF-β; growth suppressor | Reduced in aggressive cancers |
| RB | G1/S checkpoint "pocket protein" | Lost in retinoblastoma, many cancers |
| p53 | Stress sensor; DNA damage checkpoint | Most commonly mutated gene in cancer |
5. Evasion of Apoptosis
Cancer cells circumvent programmed cell death through:
- BCL-2 overexpression: t(14;18) in follicular lymphoma - BCL2 gene juxtaposed to IgH enhancer → overexpressed → blocks cytochrome c release → apoptosis inhibited
- Loss of TP53: removes the apoptotic response to DNA damage
- Activation of survival signals: PI3K/AKT axis phosphorylates and inactivates pro-apoptotic proteins like BAD
6. Growth-Promoting Metabolic Alterations (Warburg Effect / Oncometabolism)
Even with ample oxygen, cancer cells prefer aerobic glycolysis (Warburg effect), generating lactate rather than using oxidative phosphorylation. This supports:
- Rapid ATP production
- Biosynthetic intermediates for macromolecule synthesis
Oncometabolism (IDH mutations): Mutated isocitrate dehydrogenase (IDH1/IDH2) produces 2-hydroxyglutarate (2-HG), a prototypical oncometabolite. 2-HG inhibits TET2 and other α-ketoglutarate-dependent enzymes → abnormal DNA methylation → misexpression of cancer genes. IDH mutations occur in gliomas, AML, cholangiocarcinomas, and sarcomas. Targeted IDH inhibitors have been developed for clinical use.
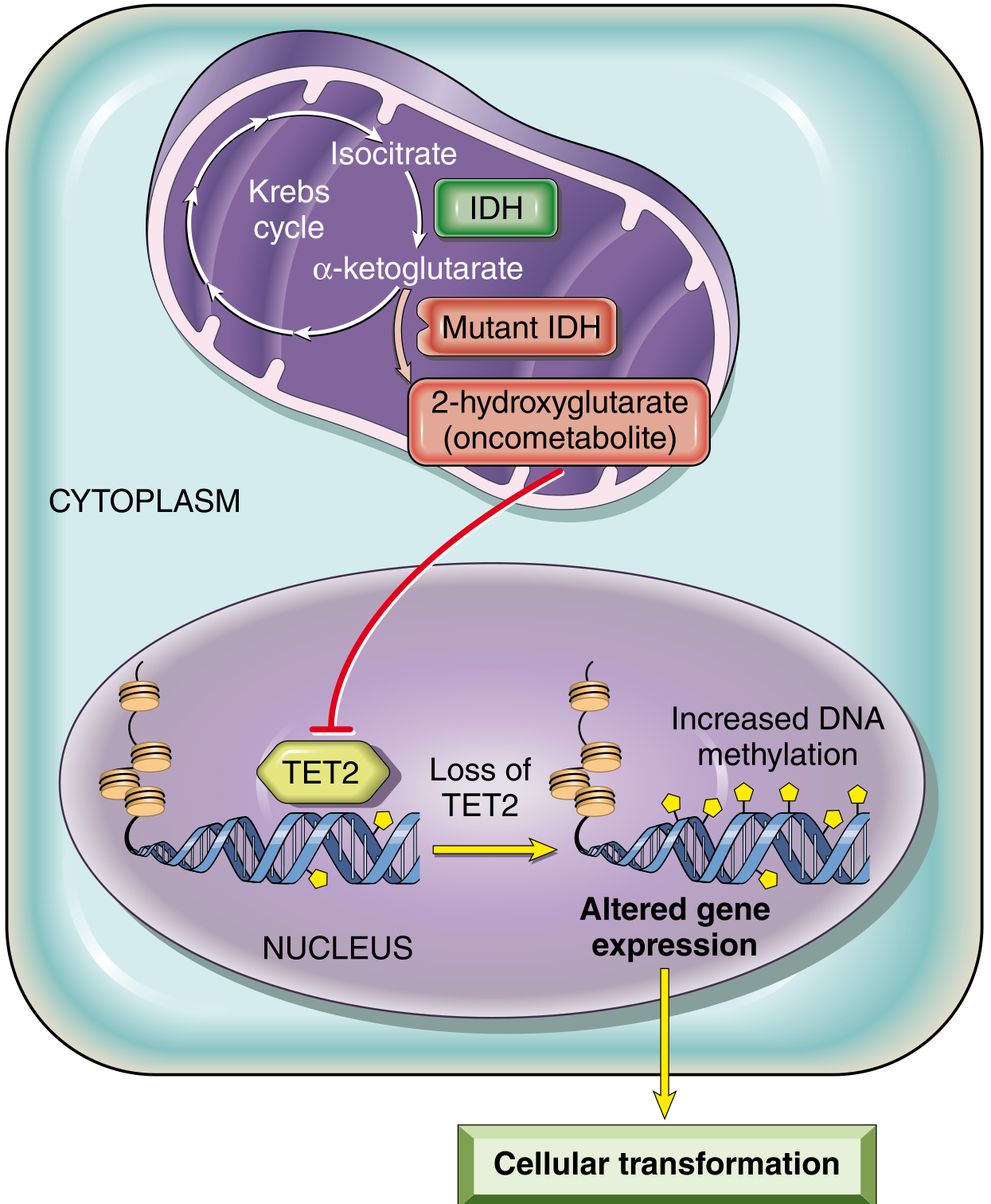
7. Limitless Replicative Potential (Telomeres and Cancer Stem Cells)
Normal cells have finite replication capacity due to telomere shortening with each division. Cancer cells escape this limit by:
- Upregulating telomerase - an enzyme that rebuilds telomere ends
- Acquiring cancer stem cell properties (self-renewal capacity), which can arise either from transformed tissue stem cells or from progenitors that acquire a mutation conferring "stemness"
8. Angiogenesis
Tumors >1-2 mm require new blood vessels (neoangiogenesis). The angiogenic switch tips the balance toward pro-angiogenic factors:
- Hypoxia → HIF-1α stabilization → VEGF and bFGF upregulation
- Loss of p53 → reduced thrombospondin-1 (anti-angiogenic)
- RAS/MYC gain-of-function → VEGF upregulation
- Proteases release ECM-stored bFGF; tumor-associated macrophages also secrete VEGF
This forms the rationale for anti-VEGF therapy (e.g., bevacizumab).
9. Invasion and Metastasis
Invasion and metastasis occur in sequential steps:
- Loosening of cell-cell contacts: Loss of E-cadherin (CDH1) expression - by promoter methylation, mutations, or transcriptional repression by SNAIL/TWIST
- Degradation of extracellular matrix: Matrix metalloproteinases (MMPs) and cathepsins degrade basement membrane and interstitial matrix; also release growth factors from ECM
- Attachment to novel ECM components: Altered integrin expression allows adhesion to new stromal sites
- Migration: Proteases and cytokines (TNF, EGF) stimulate motility
Epithelial-mesenchymal transition (EMT): Transcription factors TWIST and SNAIL drive loss of epithelial characteristics and gain of mesenchymal invasive properties.
Organ tropism of metastasis:
- Many tumors arrest in the first capillary bed encountered (lung, liver most common)
- Some show specific tropism via chemokine receptor-ligand matching with endothelial cells at target sites
- Colon → liver (via portal blood); Breast → bone, lung, brain; Prostate → bone
10. Evasion of Immune Surveillance
The immune system normally recognizes and destroys nascent cancer cells. Tumors that persist have evolved immune evasion:
- Downregulation of MHC class I molecules → invisible to CD8+ CTLs
- Expression of PD-L1 → binds PD-1 on T cells → T cell anergy (checkpoint inhibitor drugs target this)
- TGF-β secretion → immunosuppression; recruits regulatory T cells (Tregs)
- Loss of tumor antigens → clonal selection removes immunogenic variants
Tumor antigens include: products of mutated oncogenes/TSGs, overexpressed proteins, cancer-testis antigens, and viral antigens (in virus-associated cancers).
11. Genomic Instability
Cancer cells accumulate mutations far faster than normal cells, due to:
- DNA mismatch repair (MMR) defects: e.g., MLH1, MSH2 mutations in hereditary non-polyposis colorectal cancer (Lynch syndrome) → microsatellite instability (MSI)
- Nucleotide excision repair (NER) defects: e.g., XPC, XPA mutations in xeroderma pigmentosum → sensitivity to UV-induced pyrimidine dimers
- Chromosomal instability: Defects in spindle checkpoint genes → aneuploidy
12. Epigenetic Changes and Non-coding RNAs
Beyond DNA mutations:
- DNA hypermethylation of CpG islands in promoters of tumor suppressor genes (e.g., silencing RB, BRCA1, MLH1) → gene silencing without mutation
- Global DNA hypomethylation → activation of proto-oncogenes and genomic instability
- Histone modifications: Altered acetylation/methylation patterns
- microRNAs: Can act as oncogenes (OncomiRs, e.g., miR-21 suppresses PTEN) or tumor suppressors (e.g., miR-34a represses BCL2/MYC); frequently dysregulated in cancer
13. Multistep Carcinogenesis: The Colorectal Cancer Model
The classic illustration of stepwise carcinogenesis:
Normal epithelium → Hyperplasia → Early adenoma → Intermediate adenoma → Late adenoma → Carcinoma → Metastasis
Each step adds a new mutation:
- Loss of APC (tumor suppressor; Wnt pathway) - early, initiating event
- Activation of RAS - mid-progression
- Loss of SMAD4 (TGF-β pathway)
- Loss of TP53 - late event, associated with frank malignancy
This model demonstrates that individual mutations are insufficient - multiple complementary hits in different pathways are required for full malignant transformation.
14. Carcinogenic Agents
Chemical Carcinogenesis
Direct-acting carcinogens: Chemically reactive electrophiles that directly damage DNA without metabolic activation
- Examples: alkylating agents (β-propiolactone, dimethyl sulfate), nitrogen mustards, anticancer drugs (cyclophosphamide, chlorambucil)
Indirect-acting carcinogens (procarcinogens): Require metabolic activation by cytochrome P-450 monooxygenases
- Polycyclic aromatic hydrocarbons (benzo[a]pyrene from tobacco combustion) → epoxides → DNA adducts → mutations in RAS, TP53
- Aromatic amines (β-naphthylamine) → bladder cancer in dye/rubber workers
- Aflatoxin B1 (Aspergillus on stored grains) → characteristic TP53 codon 249 mutation → hepatocellular carcinoma
- Nitrosamines (from food preservatives)
Initiation vs. Promotion:
- Initiator: Causes permanent DNA mutation (single exposure sufficient); by itself insufficient for cancer
- Promoter: Stimulates proliferation of initiated cells (not mutagenic alone; requires repeated exposure AFTER initiation)
- Examples of promoters: phorbol esters, hormones, phenols
- The key action of promoters = clonal expansion of initiated cells → accumulation of additional mutations
Radiation Carcinogenesis
- UV rays (UVB, 280-320 nm): Form pyrimidine dimers (thymidine-thymidine); bypass by error-prone translesion polymerases → characteristic C→T transitions; cause skin cancers (BCC, SCC, melanoma)
- Ionizing radiation (X-rays, gamma rays): Double-strand DNA breaks → chromosomal translocations, deletions → leukemias, thyroid, breast, lung cancer; risk is dose-dependent
Microbial Carcinogenesis
Oncogenic DNA Viruses:
| Virus | Mechanism | Associated Cancer |
|---|---|---|
| HPV (high-risk: 16, 18) | E6 binds/degrades p53; E7 binds/inactivates RB | Cervical, oropharyngeal, anal cancer |
| EBV | LMP1 activates NF-κB; EBNA2 activates MYC | Burkitt lymphoma, Hodgkin lymphoma, nasopharyngeal carcinoma |
| HBV | HBx protein inhibits p53; insertional mutagenesis | Hepatocellular carcinoma |
| HHV-8 (KSHV) | Encodes viral cyclin, anti-apoptotic proteins | Kaposi sarcoma, PEL |
| Merkel cell polyomavirus | T antigen inactivates RB | Merkel cell carcinoma |
Oncogenic RNA Viruses:
- HTLV-1: Tax protein activates NF-κB, cyclins → Adult T-cell leukemia/lymphoma
Bacterial:
- H. pylori: Chronic inflammation → IL-8, ROS → mutations; CagA oncoprotein activates RAS pathway → gastric adenocarcinoma and MALT lymphoma
15. Cancer-Enabling Inflammation
The tumor microenvironment is shaped by chronic inflammation:
- Infiltrating macrophages, neutrophils, and stromal cells release EGF, VEGF, proteases, TGF-β
- Effects: promote proliferation, remove growth suppressors, enhance angiogenesis, enable invasion, suppress anti-tumor immunity, cause EMT
- This is why chronic inflammatory conditions predispose to cancer (e.g., Barrett esophagus → esophageal adenocarcinoma; ulcerative colitis → colorectal cancer)
Summary: The Hallmarks of Cancer (Hanahan & Weinberg)
| Hallmark | Key Molecular Mechanism |
|---|---|
| Self-sufficiency in growth signals | Oncogene activation (RAS, EGFR, MYC) |
| Insensitivity to growth inhibition | Tumor suppressor loss (RB, TP53, PTEN) |
| Evasion of apoptosis | BCL-2 overexpression; p53 loss |
| Limitless replicative potential | Telomerase activation; cancer stem cells |
| Angiogenesis | VEGF upregulation; HIF-1α; loss of p53 |
| Invasion and metastasis | E-cadherin loss; MMP activation; EMT |
| Altered cellular metabolism | Warburg effect; IDH mutations/oncometabolites |
| Evasion of immune surveillance | PD-L1; MHC loss; TGF-β; Tregs |
| Genomic instability | MMR defects; NER defects; chromosomal instability |
| Tumor-promoting inflammation | Macrophages, stromal cells in tumor microenvironment |
Source: Robbins, Cotran & Kumar Pathologic Basis of Disease; Robbins & Kumar Basic Pathology
Carcinogenic agent
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
I now have comprehensive material from Robbins Pathologic Basis of Disease and Robbins Basic Pathology. Let me compile the full, detailed answer.
Carcinogenic Agents
Carcinogenic agents inflict genetic damage - the fundamental driver of carcinogenesis. Three major classes have been identified: (1) Chemical agents, (2) Radiant energy (Radiation), and (3) Microbial agents (viruses and bacteria). These may act alone or in concert, sequentially producing the multiple genetic abnormalities characteristic of neoplastic cells.
- Robbins & Kumar Basic Pathology
I. CHEMICAL CARCINOGENESIS
Historical Background
Over 200 years ago, the London surgeon Sir Percival Pott correctly attributed scrotal skin cancer in chimney sweeps to chronic exposure to soot. Following this, the Danish Chimney Sweeps Guild mandated daily bathing, and scrotal cancer disappeared - one of the earliest and most effective public health measures in oncology. Since then, hundreds of chemicals have been shown to be carcinogenic.
Mechanism of Chemical Carcinogens
All direct and ultimate chemical carcinogens contain highly reactive electrophile groups that form covalent adducts with DNA, RNA, and proteins. The resulting mutations in key cancer genes - particularly RAS and TP53 - drive malignant transformation. Specific "mutational signatures" exist for different carcinogens (e.g., aflatoxin B1 causes a characteristic TP53 codon 249 mutation), which can be used as forensic tools in epidemiologic studies.
Classification: Direct vs. Indirect-Acting
A. Direct-Acting Carcinogens
- Do not require metabolic conversion to be carcinogenic
- Generally weak carcinogens
- Clinically important because some are used as chemotherapy drugs (e.g., alkylating agents used for Hodgkin lymphoma), which can later cause secondary leukemia
| Class | Examples |
|---|---|
| Alkylating agents | β-Propiolactone, dimethyl sulfate, diepoxybutane, cyclophosphamide, chlorambucil, nitrosoureas |
| Acylating agents | 1-Acetyl-imidazole, dimethylcarbamoyl chloride |
B. Indirect-Acting Carcinogens (Procarcinogens)
- Require metabolic activation (by endogenous enzymes, particularly cytochrome P-450-dependent monooxygenases) to produce the "ultimate carcinogen"
- Genetic polymorphisms in P-450 isoforms affect individual cancer risk (e.g., different CYP isoforms convert benzo[a]pyrene to carcinogenic metabolites at different rates - affecting lung cancer risk in smokers)
| Class | Key Examples | Mechanism | Associated Cancer |
|---|---|---|---|
| Polycyclic aromatic hydrocarbons | Benzo[a]pyrene (from tobacco combustion, broiled meats, smoked fish) | Metabolized to epoxides → DNA adducts | Lung cancer |
| Aromatic amines & azo dyes | β-Naphthylamine (aniline dye/rubber industry) | P-450 activation → bladder-specific carcinogens | Bladder cancer (50× increased risk) |
| Aflatoxin B1 | Produced by Aspergillus on improperly stored grains/nuts | Characteristic TP53 codon 249 mutation | Hepatocellular carcinoma (Africa, SE Asia) |
| Nitrosamines | From nitrites (food preservatives) + amines in food | Nitrosylation → alkylating intermediates | GI cancers |
| Other occupational/environmental | Vinyl chloride, arsenic, nickel, chromium, PCBs, insecticides | Various DNA damage mechanisms | Liver angiosarcoma (vinyl chloride), lung cancers |
Initiation-Promotion Sequence
A critical concept in chemical carcinogenesis is the two-stage model:
Initiation (carcinogen/mutagen):
- Single exposure sufficient
- Causes permanent, irreversible DNA mutation
- By itself insufficient to produce cancer
- Effect is heritable and cumulative
Promotion (promoter):
- NOT mutagenic or tumorigenic alone
- Requires repeated, sustained exposure AFTER initiation
- Causes clonal expansion of initiated (mutated) cells
- Stimulates proliferation → accumulation of additional mutations
- Effect is potentially reversible if exposure stops
Examples of promoters: phorbol esters, hormones (e.g., estrogen), phenols, certain drugs, conditions of tissue repair
Mechanism of promotion: Induction of cell proliferation is the sine qua non of tumor promotion. If the initiator mutated RAS, promoters drive clonal expansion of that RAS-mutant clone, which then acquires additional mutations until full malignancy is achieved.
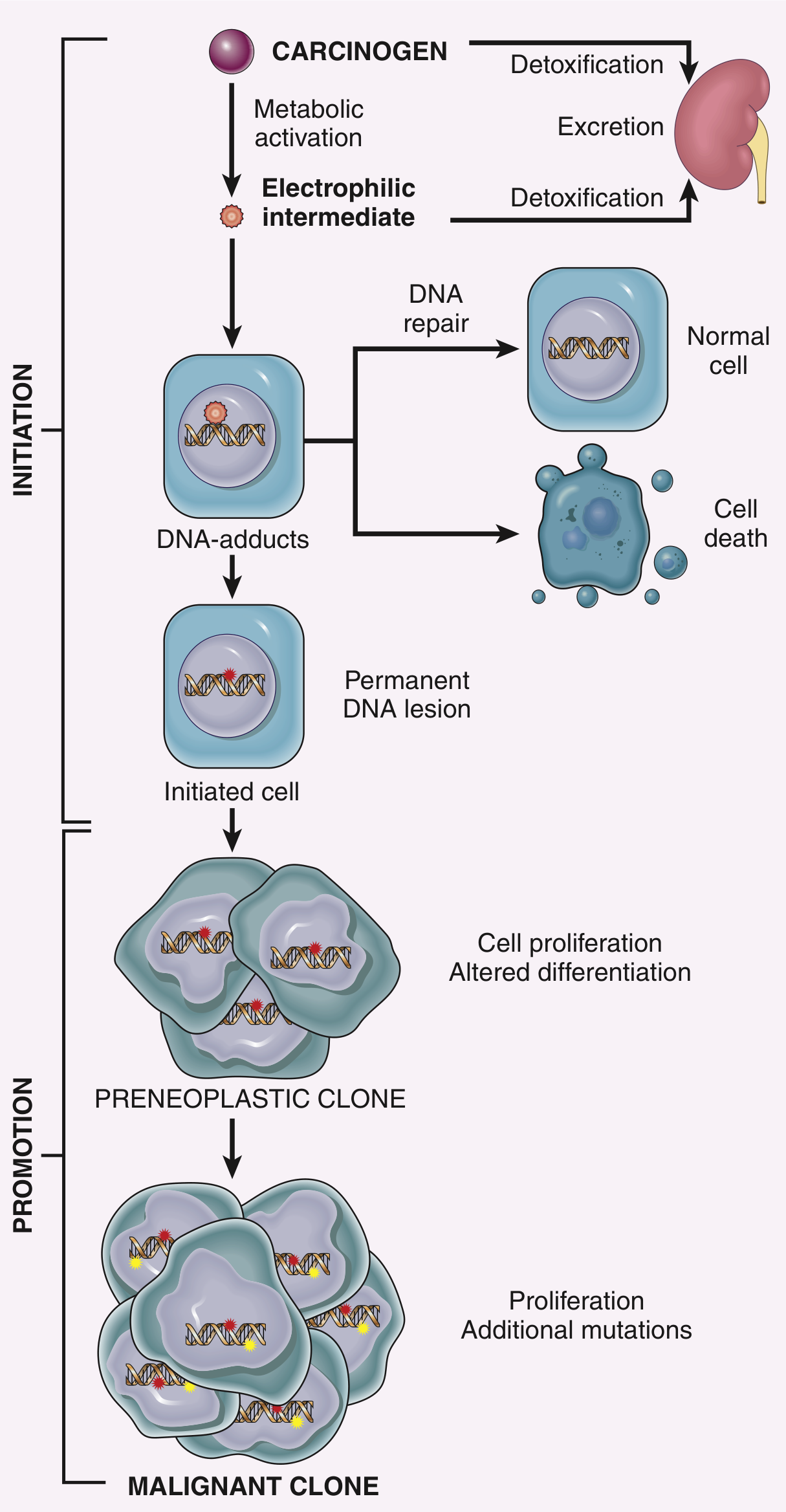
II. RADIATION CARCINOGENESIS
Radiant energy - whether UV rays of sunlight, x-rays, gamma rays, or particulate radiation - is mutagenic and carcinogenic. Radiation-induced cancers may arise decades after exposure, necessitating long observation periods.
A. Ultraviolet (UV) Radiation
Wavelength ranges:
- UVA (320-400 nm): Less carcinogenic
- UVB (280-320 nm): Most carcinogenic - causes skin cancers
- UVC (200-280 nm): Potent mutagen but filtered by the ozone layer (hence ozone depletion concerns)
Mechanism:
- UVB photons are absorbed by DNA
- Covalent cross-linking of adjacent pyrimidine bases (especially thymidine-thymidine dimers) in the same DNA strand
- DNA helix is distorted; proper base-pairing prevented
- Normally repaired by nucleotide excision repair (NER) pathway (30+ proteins)
- With excessive sun exposure, NER is overwhelmed → error-prone bypass mechanisms → characteristic C→T and CC→TT transitions (UV mutational signature)
- Key mutations in TP53 and other genes → skin cancer
Associated cancers: Squamous cell carcinoma, basal cell carcinoma, melanoma of skin
Risk factors: Fair skin, intense/cumulative sun exposure, geographic proximity to equator (e.g., Queensland, Australia has highest skin cancer rate worldwide)
- Nonmelanoma skin cancers: associated with total cumulative UV exposure
- Melanomas: associated with intense intermittent exposure (sunbathing, tanning beds)
Xeroderma Pigmentosum: Inherited defect in NER → catastrophically increased risk of UV-induced skin cancers; illustrates the protective role of DNA repair in carcinogenesis
B. Ionizing Radiation
Types: Electromagnetic (x-rays, γ-rays) AND particulate (α particles, β particles, protons, neutrons) - all carcinogenic
Mechanism:
- Causes single- and double-stranded DNA breaks (double-stranded breaks are most mutagenic)
- DNA cross-linkage with itself and with proteins
- Chromosome breakage, translocations, inversions, deletions
- Less commonly: point mutations
Historical evidence:
- X-ray pioneers developed skin cancers
- Uranium/radioactive element miners: 10× higher incidence of lung cancer
- Atomic bomb survivors (Hiroshima & Nagasaki): markedly increased leukemia (average latent period ~7 years), then thyroid, breast, colon, lung carcinomas after longer latency
- Chernobyl nuclear accident: ongoing elevated cancer incidence in surrounding populations
- Head and neck therapeutic irradiation → papillary thyroid cancers years later
Tissue vulnerability hierarchy (most to least sensitive):
- Myeloid leukemias (most frequent)
- Thyroid cancer (especially in young patients)
- Intermediate: breast, lung, salivary gland cancers
- Skin and GI epithelium (relatively resistant to radiation-induced cancer, though sensitive to acute cell killing)
CT scan risk: Children who receive 2-3 CT scans have a 3× higher risk of leukemia; 5-10 CT scans → 3× higher risk of brain tumors. Absolute risk remains low (~1 excess leukemia and 1 excess brain tumor per 10,000 CT scans over 10 years), but emphasizes minimizing unnecessary radiation exposure.
III. MICROBIAL CARCINOGENESIS
Collectively, viruses - particularly HPV, EBV, HBV, and HCV - are associated with 15-20% of cancers worldwide. A common theme: infection triggers initial polyclonal cell proliferation, which over time becomes monoclonal as driver mutations accumulate in rapidly dividing cells.
A. Oncogenic RNA Viruses
HTLV-1 (Human T-Cell Leukemia Virus Type 1)
- Only human retrovirus firmly implicated in cancer
- Endemic in: Japan, Caribbean basin, South America, Africa
- Target cell: CD4+ T cells (tropism similar to HIV)
- Transmission: Sexual intercourse, blood products, breastfeeding
- Associated cancer: Adult T-cell leukemia/lymphoma (ATLL)
- Only 3-5% of infected individuals develop ATLL, typically after 40-60 years of latency
Mechanism: HTLV-1 encodes Tax and HBZ proteins:
- Tax stimulates viral RNA transcription; activates NF-κB and other signaling pathways
- Together they promote cell proliferation, induce genomic instability, inhibit senescence
- Does NOT contain a recognizable oncogene; does NOT integrate next to a proto-oncogene
- Proviral integration is clonal, confirming infection preceded transformation
B. Oncogenic DNA Viruses
1. Human Papillomavirus (HPV)
Strains:
- Low-risk (HPV 6, 11): Cause benign squamous papillomas (warts) - low malignant potential
- High-risk (HPV 16, 18, 31): Cause cervical, anogenital, and oropharyngeal (tonsillar) cancers
Mechanism of integration: In benign warts, HPV exists as non-integrated episomal DNA. In cancers, the viral genome integrates into host chromosomes - always interrupting the E1/E2 open reading frame → loss of E2 repressor → overexpression of E6 and E7 oncoproteins
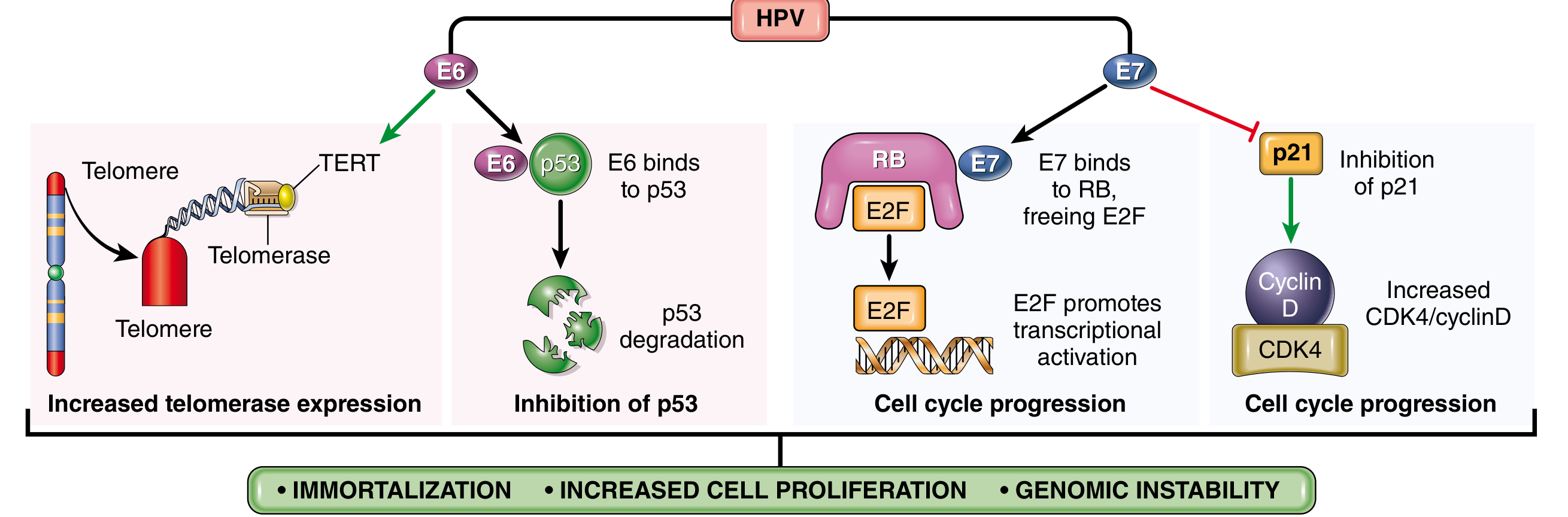
Oncogenic activities of E6:
- Binds and mediates ubiquitin-mediated degradation of p53 → abolishes DNA damage checkpoint and apoptosis
- Stimulates expression of TERT (telomerase reverse transcriptase) → cell immortalization
- High-risk HPV E6 has higher affinity for p53 than low-risk E6
Oncogenic activities of E7:
- Binds RB → displaces E2F transcription factors → constitutive G1→S progression
- Inactivates CDK inhibitors p21 and p27
- Binds and activates cyclins A and E (high-risk types 16, 18, 31)
- High-risk HPV E7 has higher affinity for RB than low-risk E7
Net effect of E6 + E7: Immortalize cells + remove restraints on proliferation + resist apoptosis → full oncogenic transformation (with additional host mutations in RAS, etc.)
Clinical implications: HPV vaccines (targeting HPV 16, 18) are highly effective in preventing cervical cancer. Women co-infected with high-risk HPV and HIV are at particularly high risk.
2. Epstein-Barr Virus (EBV)
- First virus linked to a human tumor (Burkitt lymphoma)
- Receptor: Uses complement receptor CD21 to infect B cells
- In vitro: Causes polyclonal B-cell proliferation and immortalization of B lymphoblastoid cell lines
Oncogenic proteins:
- LMP1 (Latent Membrane Protein 1): Acts as constitutively active CD40 mimic → activates NF-κB and JAK/STAT pathways → B-cell proliferation and survival
- EBNA2: Activates cyclin D and SRC-family proto-oncogenes → promotes B-cell growth
In immunocompetent individuals: CTLs eliminate EBV-infected B cells, causing self-limited infectious mononucleosis. Residual B cells downregulate immunogenic proteins and persist as long-lived memory B cells.
How Burkitt lymphoma arises (endemic): Co-infections (malaria) impair immune competence → sustained EBV-driven B-cell proliferation → CTLs eliminate most cells → small residual population acquires t(8;14) MYC translocation → monoclonal Burkitt lymphoma
| Cancer | Mechanism |
|---|---|
| Burkitt lymphoma (endemic) | EBV + malaria immunosuppression + MYC translocation |
| Nasopharyngeal carcinoma | EBV in virtually all cases |
| B-cell lymphomas (transplant/HIV patients) | EBV-driven polyclonal proliferation without T-cell control |
| Hodgkin lymphoma (subset) | EBV in Reed-Sternberg cells |
| Gastric carcinoma (subset) | EBV-positive |
3. Hepatitis B Virus (HBV) and Hepatitis C Virus (HCV)
- Together account for 70-85% of hepatocellular carcinomas worldwide
- Dominant mechanism: Immunologically mediated chronic inflammation → hepatocellular injury → reparative hepatocyte proliferation → accumulation of driver mutations
- HBx protein (HBV): Can inhibit p53; activates signal transduction pathways; promotes HIF-1α activity
- HCV core protein: Activates signal transduction pathways contributing to carcinogenesis
4. Merkel Cell Polyomavirus
- Associated with Merkel cell carcinoma (rare but aggressive skin cancer)
- T antigen inactivates RB
5. Human Herpesvirus 8 (HHV-8 / KSHV - Kaposi Sarcoma Herpesvirus)
- Encodes a viral cyclin (activates CDKs)
- Encodes anti-apoptotic proteins
- Associated with: Kaposi sarcoma, primary effusion lymphoma (PEL), Castleman disease
C. Bacterial Carcinogenesis
Helicobacter pylori
- Associated cancers: Gastric adenocarcinoma AND gastric MALToma (mucosa-associated lymphoid tissue lymphoma)
Mechanism - Multifactorial:
- Chronic inflammation → reactive oxygen species (ROS) → DNA damage in gastric epithelium
- Reparative gastric cell proliferation → accumulation of mutations
- CagA (Cytotoxin-Associated Gene A) oncoprotein: Activates RAS/MAP kinase growth factor pathways; disrupts E-cadherin
- Polyclonal B-cell proliferation in gastric mucosa → with acquisition of driver mutations → monoclonal gastric B-cell lymphoma (MALToma)
- Eradication of H. pylori with antibiotics can cause regression of low-grade MALToma (before genetic independence is acquired)
Summary Table: Carcinogenic Agents
| Agent | Mechanism | Target Gene/Pathway | Associated Cancer |
|---|---|---|---|
| Alkylating agents | Direct DNA alkylation | Any gene | Secondary leukemia |
| Benzo[a]pyrene | Epoxide-DNA adducts | RAS, TP53 | Lung cancer |
| β-Naphthylamine | P-450 activation → bladder carcinogens | - | Bladder cancer |
| Aflatoxin B1 | TP53 codon 249 mutation | TP53 | Hepatocellular carcinoma |
| UVB radiation | Pyrimidine dimers → NER overwhelmed | TP53 | Skin cancers, melanoma |
| Ionizing radiation | DNA double-strand breaks, translocations | Multiple | Leukemia, thyroid, breast, lung |
| HTLV-1 | Tax/HBZ → NF-κB, genomic instability | Multiple | Adult T-cell leukemia/lymphoma |
| HPV 16/18 | E6 degrades p53; E7 inactivates RB | TP53, RB | Cervical, oropharyngeal cancer |
| EBV | LMP1 activates NF-κB; EBNA2 activates MYC | MYC, NF-κB | Burkitt lymphoma, nasopharyngeal carcinoma |
| HBV/HCV | Chronic inflammation + HBx signaling | Multiple | Hepatocellular carcinoma |
| HHV-8 | Viral cyclin, anti-apoptotic proteins | CDKs, apoptosis | Kaposi sarcoma |
| H. pylori | Chronic inflammation + CagA → RAS | RAS, E-cadherin | Gastric adenocarcinoma, MALToma |
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease; Robbins & Kumar Basic Pathology
This is a shared conversation. Sign in to Orris to start your own chat.