Conns syndrome
Reading File
Finding Sources
Searching PubMed
"primary aldosteronism" OR "Conn syndrome"
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Here is a thorough overview of Conn's syndrome drawn from multiple authoritative textbooks.
Conn's Syndrome (Primary Hyperaldosteronism)
Definition
First described by Jerome Conn in 1955, Conn's syndrome in the strict sense refers to primary hyperaldosteronism (PA) caused by a solitary aldosterone-producing adenoma (APA). Broadly, "primary hyperaldosteronism" covers all conditions with autonomous aldosterone overproduction, suppressed renin, and resultant hypertension. It is the most common form of endocrine (secondary) hypertension.
Pathophysiology
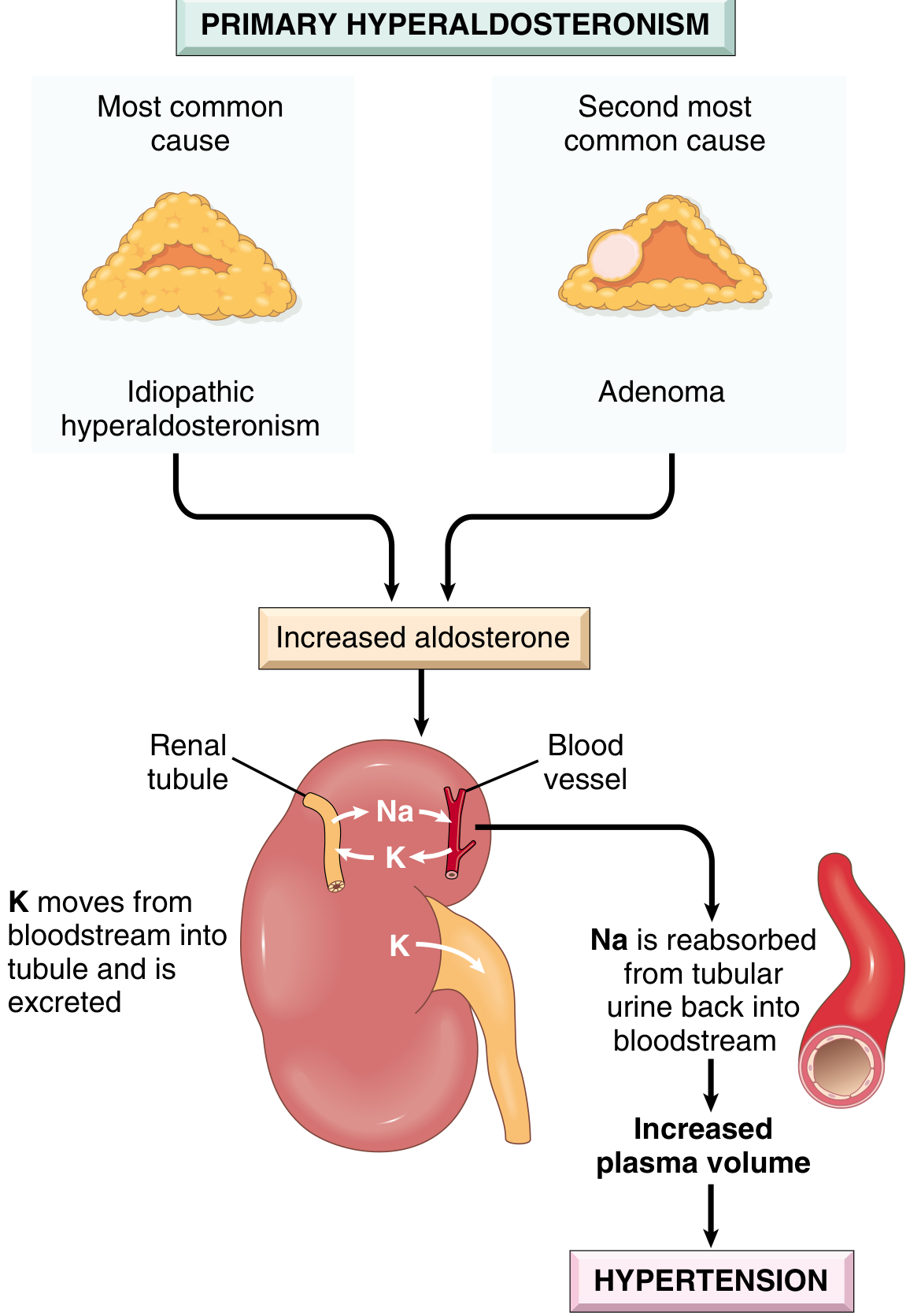
Excess aldosterone acts on the renal tubule to:
- Retain sodium → increased plasma volume → hypertension
- Excrete potassium → hypokalaemia (though many patients are normokalemic today)
- Suppress the renin-angiotensin system → low plasma renin activity (PRA)
Causes and Subtypes
| Subtype | Frequency | Key Feature |
|---|---|---|
| Bilateral idiopathic hyperplasia (BIH) | ~60% | Diffuse/nodular zona glomerulosa hyperplasia; older patients, milder HTN |
| Aldosterone-producing adenoma (APA / Conn's) | ~35% | Solitary, small (<2 cm), golden-yellow "canary tumour"; female predominance 2:1 |
| Familial hyperaldosteronism (FH-I to FH-IV) | ~5% | Autosomal dominant; FH-I (GRA) is ACTH-driven, glucocorticoid-suppressible |
| Adrenocortical carcinoma | Rare (<1%) | >4 cm, heterogeneous, irregular; very rare worldwide |
| Ectopic aldosterone-producing tumour | <0.1% | Outside the adrenal gland |
Molecular Genetics of APAs
Up to 90% of APAs harbour somatic mutations:
- KCNJ5 (~40-50%): most common; encodes GIRK4 potassium channel; loss-of-selectivity → Na influx → chronic depolarisation → autonomous aldosterone synthesis; more common in females, larger tumours
- ATP1A1, ATP2B3, CACNA1D, CTNNB1: less common variants; stronger male preponderance
Familial hyperaldosteronism:
- FH-I (GRA): CYP11B1/CYP11B2 chimeric gene on chromosome 8 → ACTH drives aldosterone synthase → treat with low-dose glucocorticoid (dexamethasone/prednisone)
- FH-II (CLCN2), FH-III (KCNJ5 germline), FH-IV (CACNA1H): autosomal dominant, early-onset PA
Clinical Features
Symptoms (often minimal):
- Hypertension (may be resistant to multiple agents)
- Muscle weakness, cramps, fatigue (if hypokalaemic)
- Polyuria and polydipsia (hypokalaemia-induced nephrogenic diabetes insipidus)
- Palpitations
Signs:
- Hypertension ± retinopathy ± bruits
- Hypokalemia (only ~1/3 of patients at presentation; normokalemia is now common)
- Metabolic alkalosis
Associations and extra risks: Elevated cardiovascular morbidity (stroke, MI, atrial fibrillation), glucose intolerance/type 2 diabetes, metabolic syndrome - all higher than matched essential hypertensives.
When to screen:
- Drug-resistant hypertension
- Hypertension + spontaneous or diuretic-provoked hypokalemia
- Hypertension + adrenal incidentaloma
- Hypertension + obstructive sleep apnoea
- Hypertension + first-degree relative with PA
- Family history of early-onset hypertension or stroke before age 40
Diagnosis
Step 1: Screening - Aldosterone-to-Renin Ratio (ARR)
- Measure plasma aldosterone concentration (PAC) and plasma renin activity (PRA) in potassium-repleted patients
- ARR >30 (PAC in ng/dL, PRA in ng/mL/h): suggestive; most common cut-off
- ARR >850 (pmol/L units) = suggestive; >1700 = highly likely
- Drugs to stop 2 weeks before: ACE inhibitors, ARBs, direct renin inhibitors, spironolactone, eplerenone
- Safe to continue: sustained-release verapamil, hydralazine, alpha-1 blockers
Step 2: Confirmatory Tests
| Test | Positive Criterion |
|---|---|
| IV saline loading (2 L over 4h) | Post-infusion PAC >10 ng/dL |
| Oral sodium loading (6 g/day x 3-5 days) | 24h urinary aldosterone >12-14 mcg/day |
| Fludrocortisone suppression | Upright PAC >6 ng/dL after 4 days of fludrocortisone 0.1 mg q6h |
| Captopril challenge | PAC remains elevated 1-2h after 25-50 mg captopril |
Step 3: Subtype Localisation
- CT adrenals (2-3 mm slices): initial investigation; APA = hypodense (<10 HU), <2 cm unilateral nodule; carcinoma = >6 cm, heterogeneous; BIH = bilateral bulky enlargement or normal
- CT has only ~78% sensitivity / 75% specificity for unilateral disease
- Adrenal vein sampling (AVS): gold standard (sensitivity 95%, specificity 100%); recommended for most surgical candidates >35 years; 4:1 cortisol-corrected aldosterone lateralisation ratio = positive
- [¹¹C]-Metomidate PET: useful adjunct when AVS fails or is equivocal (86% specific, 76% sensitive)
- Patients <35 years with a single typical hypodense nodule may proceed directly to adrenalectomy
- Genetic testing: recommended if diagnosis <20 years or family history of PA/early stroke
Treatment
Medical (for BIH or pre-operative optimisation)
- Spironolactone (aldosterone receptor antagonist): first-line; shown superior to eplerenone in an international RCT
- Eplerenone: more selective, fewer anti-androgen side effects; used when spironolactone not tolerated
- Correct hypokalemia before surgery
- GRA (FH-I): low-dose dexamethasone or prednisone at bedtime to suppress ACTH
Surgical (for APA / unilateral disease)
- Laparoscopic adrenalectomy: procedure of choice; shorter hospital stay, fewer complications vs. open
- Outcomes:
- Hypokalemia resolves in virtually all
- Hypertension is cured (BP <140/90 off medications) in ~50%
- Partial improvement (reduced antihypertensive requirement) in the remainder
- Better outcomes: younger age, shorter duration of hypertension, controlled on ≤2 agents pre-op, fewer hypertensive first-degree relatives
Post-operative monitoring: Check PAC and PRA shortly after surgery; discontinue potassium supplements and aldosterone antagonists; temporary IV saline may be needed while the contralateral adrenal recovers.
Recent Evidence
A 2025 Endocrine Society systematic review (PMID 40658500) underpins updated guidelines on PA management. A 2025 meta-analysis in Lancet Diabetes & Endocrinology (PMID 41235994) showed that post-treatment renin status in medically treated PA predicts cardiovascular and renal outcomes - a useful therapeutic monitoring target. A 2024 Mendelian randomisation study (PMID 39101507) confirmed a causal link between primary aldosteronism and adverse cardiovascular outcomes.
Sources: Robbins Pathologic Basis of Disease; Bailey & Love's Surgery (28th ed.); Brenner & Rector's The Kidney; Braunwald's Heart Disease; Grainger & Allison's Diagnostic Radiology
Give me a10 mark physiology answer for conns syndrome
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
I now have all the physiology-focused content needed. Here is a structured 10-mark physiology answer:
Conn's Syndrome - Physiology (10 Marks)
Definition (1 mark)
Conn's syndrome is primary hyperaldosteronism caused by a unilateral aldosterone-secreting adrenal adenoma arising in the zona glomerulosa. It results in autonomous, renin-independent excess secretion of aldosterone, leading to a predictable constellation of physiological disturbances.
Normal Physiology of Aldosterone (1 mark)
Aldosterone is the principal mineralocorticoid of the adrenal cortex. It acts on the principal cells (P cells) of the collecting duct and late distal tubule - the aldosterone-sensitive distal nephron - via intracellular mineralocorticoid receptors (MR). Its actions are:
- Promotes Na+ reabsorption from tubular lumen into the blood
- Promotes K+ and H+ secretion into the tubular lumen
- Net effect: expands extracellular fluid (ECF) volume, maintains K+ homeostasis
Aldosterone is normally regulated by the renin-angiotensin-aldosterone system (RAAS): low renal perfusion → renin release → angiotensin II → aldosterone secretion.
Mechanism of Action of Aldosterone (1 mark)
Aldosterone binds the cytoplasmic MR → receptor-hormone complex translocates to the nucleus → alters gene transcription → two effects:
- Rapid effect: increases insertion of pre-formed ENaC (epithelial Na+ channel) subunits into the apical membrane from cytoplasmic vesicles
- Slow (genomic) effect: increases synthesis of ENaC subunits (α, β, γ) and activates SGK1 (serum- and glucocorticoid-regulated kinase), which phosphorylates Nedd4-2, preventing ENaC internalisation and prolonging its membrane residence
The basolateral Na+/K+-ATPase pumps the reabsorbed Na+ out into the bloodstream, generating the electrochemical gradient that drives K+ and H+ secretion into the lumen.
Pathophysiology of Conn's Syndrome (4 marks)
In Conn's syndrome, autonomous aldosterone secretion is independent of RAAS control, producing the following cascade:
1. Sodium Retention → Volume Expansion → Hypertension
- Excess aldosterone → ↑ ENaC expression and activity → ↑ Na+ reabsorption in collecting ducts
- Na+ retention → ↑ ECF volume → ↑ plasma volume → ↑ cardiac output → hypertension
- GFR rises, proximal tubule reabsorption falls → generous distal tubular salt delivery, which further amplifies distal Na+ reabsorption and K+/H+ secretion
2. Hypokalaemia
- The increased luminal electronegativity created by Na+ reabsorption drives K+ secretion into the lumen → urinary K+ wasting → hypokalaemia
- K+ depletion causes K+ to shift out of cells in exchange for H+ → generates additional ECF HCO3-
- Hypokalaemia activates H+/K+-ATPase in type A intercalated cells → further H+ secretion
- Clinical effects of hypokalaemia: muscle weakness, cramps, fatigue, periodic paralysis, polyuria/polydipsia (nephrogenic diabetes insipidus due to impaired aquaporin regulation)
3. Metabolic Alkalosis
- Excess aldosterone drives H+ secretion into the tubular lumen (via H+-ATPase in intercalated cells) → HCO3- generation → metabolic alkalosis
- Hypokalaemia independently amplifies alkalosis (K+/H+ cell exchange, ↑ ammoniagenesis)
4. Suppressed Renin
- Volume expansion → ↑ renal perfusion pressure → suppression of renin release from the juxtaglomerular apparatus
- This is the key distinguishing feature from secondary hyperaldosteronism (where both renin AND aldosterone are elevated)
- Low plasma renin activity (PRA) combined with high aldosterone = the diagnostic hallmark (elevated aldosterone-to-renin ratio, ARR)
Summary of Physiological Disturbances (1 mark)
| Parameter | Change | Mechanism |
|---|---|---|
| Aldosterone | ↑↑ (autonomous) | Adenoma secretion |
| Plasma renin activity | ↓↓ (suppressed) | ECF volume expansion → ↓ juxtaglomerular renin |
| Sodium | ↑ (retained) | ↑ ENaC activity |
| Potassium | ↓ (hypokalaemia) | ↑ K+ secretion |
| Blood pressure | ↑ (hypertension) | ↑ ECF/plasma volume |
| Blood pH | ↑ (alkalosis) | ↑ H+ secretion |
| HCO3- | ↑ | H+ loss + K+/H+ cell shift |
"Aldosterone Escape" Phenomenon (1 mark)
Despite ongoing aldosterone excess, oedema does not usually develop. This is because the ECF volume expansion eventually triggers pressure natriuresis and suppresses proximal tubular reabsorption (via atrial natriuretic peptide and reduced angiotensin II), which "escapes" the Na+-retaining effect. However, hypokalaemia and hypertension persist because there is no equivalent escape mechanism for K+ or H+ secretion.
Treatment (Physiological Basis) (1 mark)
| Treatment | Mechanism |
|---|---|
| Spironolactone / Eplerenone | Competitively block the mineralocorticoid receptor → reduce ENaC expression → ↓ Na+ retention, ↓ K+ loss |
| Laparoscopic adrenalectomy | Removes source of autonomous aldosterone → RAAS recovers, electrolytes and BP normalise |
| Potassium supplementation | Corrects hypokalaemia pre-operatively |
| Low-salt diet | Reduces distal tubular Na+ delivery → blunts K+ and H+ secretion |
Sources: Costanzo Physiology (7th ed.); Ganong's Review of Medical Physiology (26th ed.); Comprehensive Clinical Nephrology (7th ed.); Brenner & Rector's The Kidney
This is a shared conversation. Sign in to Orris to start your own chat.