Write a very short note on urea cycle and steps from vasudevan
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Here is a concise note on the Urea Cycle in the style of Vasudevan's biochemistry:
Urea Cycle (Krebs-Henseleit Cycle)
Introduction
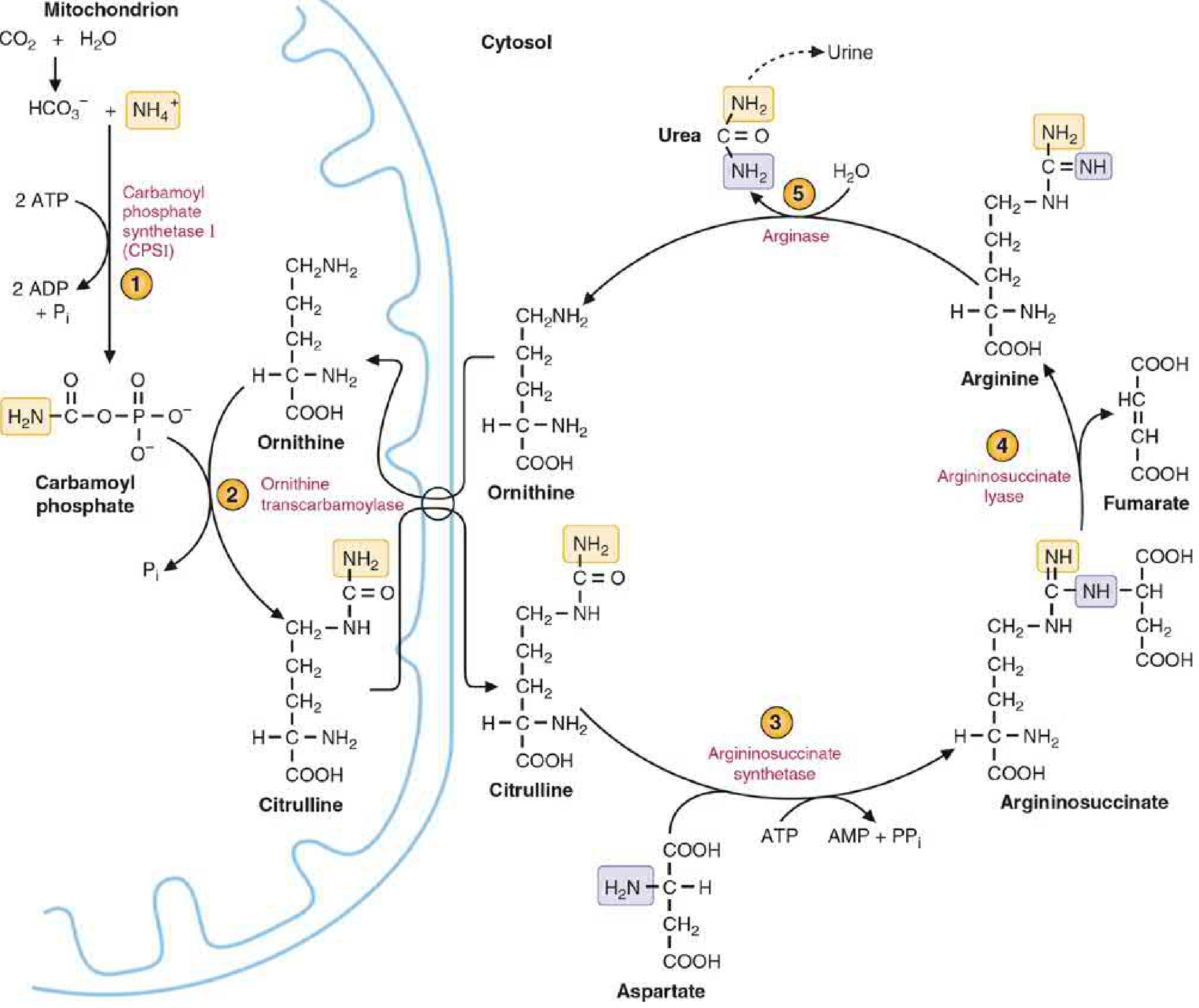
The urea cycle is the primary mechanism by which the body disposes of toxic ammonia (NH₃/NH₄⁺). It was first described in 1932 by Hans Krebs and Kurt Henseleit, hence also called the Krebs-Henseleit cycle. Urea accounts for ~90% of urinary nitrogen. The cycle occurs mainly in hepatocytes and takes place partly in the mitochondrial matrix (steps 1-2) and partly in the cytosol (steps 3-5).
- Nitrogen enters as NH₄⁺ (from amino acid catabolism) and aspartate
- Carbon and oxygen are derived from CO₂ (HCO₃⁻)
- Ornithine is the primer - it is regenerated at the end of each turn (analogous to oxaloacetate in the TCA cycle)
Steps of the Urea Cycle
Step 1 - Formation of Carbamoyl Phosphate (Mitochondria)
Enzyme: Carbamoyl Phosphate Synthetase I (CPS-I)
- NH₄⁺ + HCO₃⁻ + 2 ATP → Carbamoyl phosphate + 2 ADP + Pi
- CPS-I requires N-acetylglutamate (NAG) as an obligatory allosteric activator
- NAG is synthesized from acetyl-CoA + glutamate by NAG synthase, which is activated by arginine
- Note: CPS-II (cytosolic) participates in pyrimidine synthesis; it uses glutamine as nitrogen source and does NOT need NAG
Step 2 - Formation of Citrulline (Mitochondria → Cytosol)
Enzyme: Ornithine Transcarbamylase (OTC)
- Ornithine + Carbamoyl phosphate → Citrulline + Pi
- Citrulline is transported to the cytosol via an ornithine-citrulline antiporter in the inner mitochondrial membrane
- OTC deficiency is the most common urea cycle enzyme defect
Step 3 - Formation of Argininosuccinate (Cytosol)
Enzyme: Argininosuccinate Synthetase
- Citrulline + Aspartate + ATP → Argininosuccinate + AMP + PPi
- This step incorporates the second nitrogen (from aspartate) into the cycle
- 1 ATP is consumed (equivalent to 2 high-energy bonds as PPi is released)
Step 4 - Cleavage of Argininosuccinate (Cytosol)
Enzyme: Argininosuccinate Lyase (Argininosuccinase)
- Argininosuccinate → Arginine + Fumarate
- Fumarate links the urea cycle to the TCA cycle (converted to malate → oxaloacetate → aspartate, completing the "urea cycle-TCA cycle link")
Step 5 - Cleavage of Arginine → Urea + Ornithine (Cytosol)
Enzyme: Arginase-I (liver-specific)
- Arginine + H₂O → Urea + Ornithine
- Ornithine is transported back into the mitochondria to begin another turn
- Arginase-I is virtually exclusive to the liver - only the liver synthesizes urea
Summary Table
| Step | Location | Substrate → Product | Enzyme |
|---|---|---|---|
| 1 | Mitochondria | NH₄⁺ + HCO₃⁻ → Carbamoyl phosphate | CPS-I |
| 2 | Mitochondria | Ornithine → Citrulline | OTC |
| 3 | Cytosol | Citrulline + Aspartate → Argininosuccinate | Argininosuccinate synthetase |
| 4 | Cytosol | Argininosuccinate → Arginine + Fumarate | Argininosuccinate lyase |
| 5 | Cytosol | Arginine → Urea + Ornithine | Arginase-I |
Energy Cost
Overall stoichiometry:
NH₄⁺ + HCO₃⁻ + Aspartate + 3 ATP + H₂O → Urea + Fumarate + 2 ADP + AMP + 2Pi + PPi
- 3 ATP molecules are consumed per mole of urea formed (equivalent to 4 high-energy phosphate bonds)
- The synthesis of urea is irreversible
Key Points (Exam Focus)
-
Two nitrogens of urea: one from NH₄⁺ (via glutamate/GDH), one from aspartate (via transamination)
-
Allosteric activator of CPS-I: N-acetylglutamate (NAG) - rate-limiting regulator
-
Most common enzyme defect: OTC deficiency (X-linked)
-
Only organ synthesizing urea: Liver
-
Urea cycle-TCA cycle link: via fumarate → malate → oxaloacetate → aspartate
-
Patients with liver failure accumulate ammonia → hepatic encephalopathy
-
Biochemistry, 8th ed Lippincott Illustrated Reviews, p. 712-715
-
Basic Medical Biochemistry - A Clinical Approach, 6e, p. 1321-1322
Disorders linked to deficiency of urea cycle enzymes
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
I now have comprehensive data from multiple authoritative sources. Here is the complete note:
Disorders Linked to Deficiency of Urea Cycle Enzymes
Overview
Genetic deficiencies of each of the five urea cycle enzymes (plus NAGS) have been described. Overall incidence is approximately 1:25,000-1:35,000 live births. They are all autosomal recessive except OTC deficiency, which is X-linked and is the most common defect (~57-62% of cases). The universal consequence is hyperammonemia, which causes direct neuronal toxicity and brain edema.
Common symptoms across all defects: protein aversion, recurrent vomiting, lethargy, tremors, slurred speech, cerebral edema, intellectual/developmental disability, and in severe cases coma and death. Neonates with complete deficiency typically present at 1-4 days of life.
Individual Enzyme Defects
1. N-Acetylglutamate Synthase (NAGS) Deficiency
- Enzyme blocked: NAGS (makes the allosteric activator of CPS-I)
- Accumulated metabolite: Ammonia (no diagnostic accumulation of cycle intermediates)
- Key lab: Low citrulline, low orotic acid, elevated glutamine
- Features: Clinically indistinguishable from CPS-I deficiency; episodic hyperammonemia triggered by high-protein meals, stress, or fasting
- Treatment: Carglumic acid (N-carbamylglutamate) - a synthetic NAG analogue; this is specific to NAGS deficiency and can restore full urea cycle function, rendering other therapies unnecessary
2. Carbamoyl Phosphate Synthetase I (CPS-I) Deficiency
- Enzyme blocked: CPS-I (Step 1, mitochondria)
- Accumulated metabolite: Ammonia
- Key lab: Low/absent citrulline, low orotic acid (unlike OTC deficiency), elevated glutamine
- Features: Severe neonatal hyperammonemia; autosomal recessive
- Distinguishing point from OTC: Urine orotic acid is normal/low (carbamoyl phosphate does not escape to cytosol, so pyrimidine synthesis is not overstimulated)
3. Ornithine Transcarbamylase (OTC) Deficiency (Most Common)
- Enzyme blocked: OTC (Step 2, mitochondria)
- Accumulated metabolite: Carbamoyl phosphate, ammonia
- Key lab: ↑ Orotic acid in blood and urine, ↓ citrulline, ↓ arginine, ↑ glutamine
- Why orotic acid rises: Excess carbamoyl phosphate "overflows" into the cytosol and enters pyrimidine biosynthesis, producing excess orotic acid - this is the hallmark diagnostic marker
- Inheritance: X-linked - severely affects males; females (carriers) may be symptomatic depending on X-inactivation pattern. Can present in females at time of childbirth due to catabolism
- Incidence: Most frequent urea cycle defect (57-62%)
4. Argininosuccinate Synthetase Deficiency - Citrullinemia Type 1
- Enzyme blocked: Argininosuccinate synthetase (Step 3, cytosol)
- Accumulated metabolite: Citrulline (markedly elevated in blood and urine), orotic acid
- Key lab: Very high plasma citrulline - detectable on newborn screening
- Forms:
- Neonatal acute (classic) - severe hyperammonemia at birth
- Milder late-onset form
- Pregnancy-onset form
- Asymptomatic form
- Note: Citrin deficiency (defect in aspartate/glutamate exchanger) causes Citrullinemia Type 2 - a different mechanism but also elevated citrulline
5. Argininosuccinate Lyase (ASL) Deficiency - Argininosuccinic Aciduria
- Enzyme blocked: Argininosuccinate lyase (Step 4, cytosol)
- Accumulated metabolite: Argininosuccinate in blood and urine; milder citrulline elevation
- Key lab: Argininosuccinic acid detectable on plasma amino acid analysis and newborn screening; 2nd most common urea cycle defect (11.5-18%)
- Features: Hyperammonemia + liver cirrhosis (hepatic involvement is characteristic); neurologic abnormalities, developmental delays, cognitive impairment
- Treatment: Arginine supplementation (citrulline → argininosuccinate → arginine pathway used as "nitrogen sink")
6. Arginase-I Deficiency - Argininemia / Hyperargininemia
- Enzyme blocked: Arginase-I (Step 5, cytosol; liver-specific)
- Accumulated metabolite: Arginine (elevated in blood and urine)
- Key lab: High plasma arginine; hyperammonemia is often less severe because arginine itself contains two waste nitrogens and is excreted
- Features: Unique clinical presentation - patients appear healthy at birth and develop normally for 1-3 years, then develop progressive spastic diplegia, loss of developmental milestones, intellectual disability, seizures
- Distinguishing feature: Milder and delayed hyperammonemia compared to other urea cycle defects
Summary Comparison Table
| Enzyme Deficiency | Step | Accumulated Metabolite | Orotic Acid | Citrulline | Key Feature |
|---|---|---|---|---|---|
| NAGS | Pre-Step 1 | NH₃ | Low/normal | Low | Responds to carglumic acid |
| CPS-I | Step 1 | NH₃ | Low/normal | Low | No orotic aciduria |
| OTC | Step 2 | NH₃, carbamoyl-P | ↑↑ HIGH | Low | X-linked; most common |
| ASS (Citrullinemia) | Step 3 | Citrulline | ↑ | ↑↑ HIGH | Newborn screen (+) |
| ASL (Argininosuccinic aciduria) | Step 4 | Argininosuccinate | Normal | Mildly ↑ | Liver cirrhosis |
| Arginase-I (Argininemia) | Step 5 | Arginine | Normal | Normal | Delayed onset; mild NH₃ |
Diagnostic Key Points
- Orotic acid elevated: OTC deficiency (orotic acid is the key differentiator from CPS-I and NAGS)
- Orotic acid normal/low: CPS-I deficiency, NAGS deficiency
- High citrulline: Citrullinemia (ASS deficiency) or citrin deficiency
- Argininosuccinate in plasma/urine: ASL deficiency
- High arginine, mild NH₃, late onset: Arginase deficiency
- All defects → elevated plasma glutamine (ammonia not disposed by cycle is conjugated to glutamate by glutamine synthase in perivenous hepatocytes)
Treatment Principles
- Acute hyperammonemia: IV glucose + lipids to reverse catabolism; IV arginine (essential in these patients except arginase deficiency)
- Nitrogen scavengers:
- Sodium benzoate → conjugates glycine → hippurate (excreted in urine)
- Sodium phenylbutyrate/phenylacetate → conjugates glutamine → phenylacetylglutamine (excreted in urine)
- Hemodialysis if ammonia does not fall promptly
- Long-term: Protein-restricted diet, citrulline (for OTC/CPS-I deficiency), arginine (for ASS/ASL deficiency), phenylbutyrate/glycerol phenylbutyrate
- NAGS deficiency: Carglumic acid (synthetic NAG analogue) - specific and highly effective
- Liver transplantation for severe, medically refractory cases
- Biochemistry, 8th ed Lippincott Illustrated Reviews, p. 725-728
- Harrison's Principles of Internal Medicine 22E, p. 3415-3416
- Tietz Textbook of Laboratory Medicine, 7th Ed, p. 2474-2475
- Goldman-Cecil Medicine, Table 189-5
This is a shared conversation. Sign in to Orris to start your own chat.