Congenital adrenal hyperplasia
Reading File
Finding Sources
Searching PubMed
"congenital adrenal hyperplasia"[MeSH Terms]
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
I now have comprehensive information from multiple authoritative textbooks. Let me compile the full answer.
Congenital Adrenal Hyperplasia (CAH)
Definition
Congenital adrenal hyperplasia is a group of autosomal recessive disorders caused by inherited enzymatic defects in adrenal steroidogenesis. The fundamental defect in all forms is inadequate cortisol synthesis, which drives a compensatory increase in CRH and ACTH, leading to bilateral adrenal cortex hyperplasia and accumulation of steroid precursors proximal to the enzymatic block - many of which are shunted into androgen synthesis.
- Goldman-Cecil Medicine, p. 2515
- Creasy & Resnik's Maternal-Fetal Medicine
Steroid Biosynthesis Pathway
The following diagram illustrates normal adrenal and gonadal steroidogenesis. The key enzymes affected in CAH are shown - particularly 21-hydroxylase (CYP21A2), 11β-hydroxylase (CYP11B1), and others further upstream.
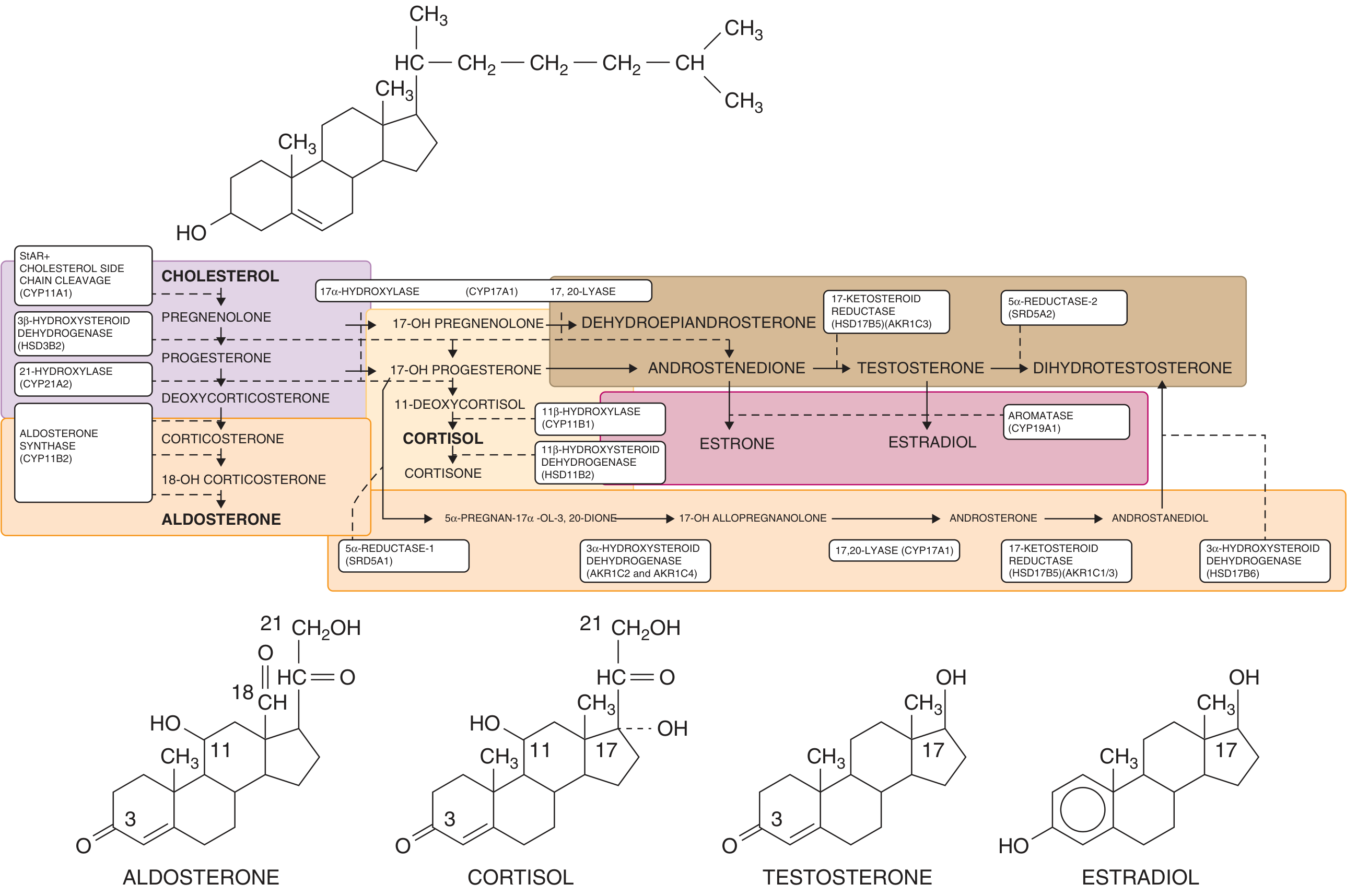
Goldman-Cecil Medicine - Adrenal and gonadal steroidogenesis
Enzymatic Defects and Relative Frequency
| Enzyme | Gene | Frequency | Key Features |
|---|---|---|---|
| 21-Hydroxylase | CYP21A2 | ~95% of all CAH | Virilization ± salt wasting |
| 11β-Hydroxylase | CYP11B1 | ~5% | Virilization + hypertension |
| 3β-HSD | HSD3B2 | Rare | Incomplete masculinization (males), mild virilization (females) |
| 17α-Hydroxylase | CYP17A1 | Rare | Hypertension, sexual infantilism |
| Cholesterol side-chain cleavage | CYP11A1 | Very rare | Fatal/incompatible with reproduction |
- Campbell-Walsh-Wein Urology; Emery's Medical Genetics
21-Hydroxylase Deficiency (Most Common Form)
Genetics
-
Gene: CYP21A2 on chromosome 6p21.3, within the HLA complex
-
Transmitted in autosomal recessive pattern
-
Adjacent pseudogene CYP21PA1 is 98% homologous - gene conversion events during meiosis are a major mutation mechanism
-
200 mutations identified; 95% of causative mutations are known
-
Incidence: 1 in 5,000-15,000 in general populations; as high as 1 in 490 in Yupik Alaskan Eskimos
-
Campbell-Walsh-Wein Urology
Pathophysiology
The 21-hydroxylase enzyme converts:
- Progesterone → Deoxycorticosterone (mineralocorticoid pathway)
- 17-OH Progesterone → 11-Deoxycortisol (glucocorticoid pathway)
When blocked, 17-OH progesterone accumulates and is shunted to androstenedione → testosterone, causing virilization. The degree of enzyme deficiency determines the clinical phenotype.
Clinical Subtypes
| Subtype | Residual Enzyme Activity | Features |
|---|---|---|
| Salt-wasting (classic) | 0-1% | Virilization + mineralocorticoid deficiency; electrolyte crisis 5-15 days after birth |
| Simple virilizing (classic) | 2-20% | Virilization without salt wasting; sufficient mineralocorticoid preserved |
| Non-classic (late-onset) | >20% | Mild: hirsutism, acne, menstrual irregularities, oligospermia; diagnosed in adolescence/adulthood |
- Robbins & Cotran Pathologic Basis of Disease; Creasy & Resnik
Salt-Wasting Crisis
The salt-wasting form presents soon after birth (5-15 days in males, often recognized at birth in females due to virilized genitalia). Features:
-
Hyponatremia, hyperkalemia, metabolic acidosis
-
Hypotension, cardiovascular collapse, potentially fatal
-
Males are often not diagnosed until the crisis occurs (normal appearing genitalia at birth)
-
Robbins & Cotran
Clinical Features by Enzyme Deficiency
In 46,XX Females (Virilization)
- In utero: Clitoral hypertrophy, labial fusion, urogenital sinus - degrees of difference in sexual development (DSD)
- Childhood: Rapid somatic growth initially, then premature epiphyseal closure → short adult stature without treatment
- Adolescence/adulthood (non-classic): Hirsutism, acne, oligomenorrhea, PCOS-like picture
In 46,XY Males
- Typically normal genitalia at birth (endogenous androgens are sufficient)
- Salt-wasting crisis in first 2 weeks is the presenting feature in severe forms
- Testicular adrenal rest tumors (TARTs) are a long-term complication - can cause severe testicular damage and infertility
- Non-classic: Oligospermia, subfertility
Adrenomedullary Dysfunction
In severe salt-wasting CAH, high intra-adrenal cortisol is needed for medullary catecholamine synthesis. Low cortisol + adrenomedullary dysplasia impairs epinephrine secretion, worsening hypotension and circulatory collapse.
- Robbins & Cotran, p. 2614-2615
Pathological Morphology (Robbins)
- Bilateral adrenal hyperplasia: glands may reach 10-15x normal weight
- Cortex is thickened and nodular, brown on cut section (lipid depletion)
- Proliferating cells: compact, eosinophilic, lipid-depleted cells intermixed with lipid-laden clear cells
- Anterior pituitary: hyperplasia of corticotroph (ACTH-producing) cells
Diagnosis
Biochemical
- 17-Hydroxyprogesterone (17-OHP): elevated in 21-hydroxylase deficiency - the key diagnostic marker
- Classic CAH: markedly elevated basally
- Non-classic: may require ACTH stimulation test; exaggerated 17-OHP rise (>1,500 ng/dL)
- Renin/aldosterone ratio: elevated plasma renin in salt-wasting forms
- Electrolytes: hyponatremia, hyperkalemia in salt-wasting crisis
- Androstenedione, testosterone: elevated
Newborn Screening
- Most countries screen for 21-hydroxylase deficiency via elevated 17-OHP on heel-prick blood spot (day 2-5 of life)
- Cannot detect non-classic CAH on routine newborn screening
Genetic Testing
-
CYP21A2 genotyping confirms diagnosis and guides prognosis
-
Prenatal diagnosis: elevated 17-OHP or 21-deoxycortisol in amniotic fluid, or molecular testing via CVS/amniocentesis
-
Berek & Novak's Gynecology; Creasy & Resnik
Treatment
Glucocorticoid Replacement
- Hydrocortisone: 10-20 mg/m² body surface area/day in divided doses (preferred in children to minimize growth suppression)
- Goal: suppress morning 17-OHP to 300-900 ng/dL, normalize androgen levels
- Overreplacement causes Cushingoid effects and impairs growth; underreplacement allows continued androgen excess and epiphyseal advancement
- In adults: prednisone or dexamethasone can be used
Mineralocorticoid Replacement
- Fludrocortisone: indicated in salt-wasting forms; many guidelines recommend it even in simple virilizing forms
- Goal: suppress plasma renin activity to <5 ng/mL/hr
- Sodium supplementation in infants
Stress Dosing
- Double or triple glucocorticoid dose during febrile illness, surgery, or trauma to prevent adrenal crisis
Monitoring
- Children: growth velocity, bone age, hormone levels (17-OHP, androstenedione)
- Adults: 17-OHP, androstenedione, renin, blood pressure, BMI
- Males: testicular ultrasound for TART surveillance
Surgical Management
-
Females with ambiguous genitalia: clitoral recession and vaginoplasty - timing is debated; most centers perform in early childhood but the decision involves multidisciplinary input and, increasingly, patient/family preference
-
A multidisciplinary team should guide sex-of-rearing decisions and any interventions
-
Berek & Novak's Gynecology; Creasy & Resnik; Campbell-Walsh-Wein Urology
Pregnancy and CAH
Women with CAH have reduced fertility due to:
- Adrenal androgens suppressing ovulation (direct ovarian effect + GnRH suppression)
- Elevated progesterone reducing LH pulse frequency and thickening cervical mucus
Management in pregnancy:
- Switch dexamethasone to hydrocortisone (dexamethasone crosses the placenta; hydrocortisone is inactivated by placental 11β-HSD)
- Target progesterone <0.6 ng/mL for optimal fertility
- Fludrocortisone continued in salt-losing forms
- 100% of offspring are carriers if the partner is unaffected; 25% of offspring are affected if both parents are carriers
Prenatal treatment with dexamethasone (to the mother) can reduce or prevent virilization of an affected female fetus, but carries maternal hypercortisolism risks and uncertain fetal neurodevelopmental effects - this is no longer routinely recommended without careful specialist oversight.
- Creasy & Resnik's Maternal-Fetal Medicine
11β-Hydroxylase Deficiency (Second Most Common)
-
~5% of CAH cases
-
Defect: CYP11B1; blocks conversion of 11-deoxycortisol → cortisol and deoxycorticosterone → corticosterone
-
Deoxycorticosterone accumulates → hypertension and hypokalemia (mineralocorticoid excess)
-
Paradoxically, aldosterone synthesis is low but DOC compensates
-
Also causes androgen excess and virilization (similar to 21-hydroxylase deficiency)
-
Campbell-Walsh-Wein Urology; Robbins & Cotran
Non-Classic CAH
- Partial 21-hydroxylase deficiency; most common form overall in the general population
- Presents in adolescence or adulthood with hirsutism, acne, oligomenorrhea, PCOS-like picture
- Cannot be identified by newborn screening
- Diagnosed by ACTH stimulation test (exaggerated 17-OHP response)
- Males: oligospermia; infertility may be reversed with glucocorticoid therapy at lower doses
- Treatment: low-dose glucocorticoids when symptomatic; OCP + anti-androgens are alternatives
Key Summary Points
- Fundamental defect: Impaired cortisol synthesis → elevated ACTH → adrenal hyperplasia + androgen excess
- Most common: 21-hydroxylase deficiency (CYP21A2), >95% of cases
- Three classic subtypes: Salt-wasting, simple virilizing, non-classic
- 46,XX: Virilized genitalia (DSD) at birth; 46,XY: normal genitalia but salt crisis at 5-15 days
- Diagnosis: Elevated 17-OHP is the key marker; confirmed by genetic testing
- Newborn screening detects classic forms
- Treatment: Hydrocortisone (glucocorticoid replacement) + fludrocortisone (mineralocorticoid) + salt supplementation in infants
- Long-term risks: Short stature, infertility, testicular adrenal rest tumors (males), metabolic syndrome
Sources: Goldman-Cecil Medicine (26th ed.), Robbins & Cotran Pathologic Basis of Disease, Campbell-Walsh-Wein Urology, Berek & Novak's Gynecology, Creasy & Resnik's Maternal-Fetal Medicine, Emery's Medical Genetics
This is a shared conversation. Sign in to Orris to start your own chat.