Chronic myelogenous leukaemia from Robbins pathology
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Loading Image
Loading Image
Loading Image
I now have comprehensive material from both Robbins texts with all key images. Here is the complete answer:
Chronic Myelogenous Leukaemia (CML) - Robbins Pathology
Overview and Classification
CML is a myeloproliferative neoplasm - a group of clonal stem cell disorders characterized by mutated, constitutively activated tyrosine kinases that confer growth factor independence. Among all myeloproliferative neoplasms, CML is set apart by one defining feature: the BCR-ABL fusion gene.
Epidemiology
- Primarily a disease of adults, peak incidence in the fifth to sixth decades of life
- Also occurs in children and adolescents
- Approximately 4,800 new cases per year in the United States
- No strong environmental risk factors identified (though prior radiation exposure is one)
Pathogenesis
The Philadelphia Chromosome
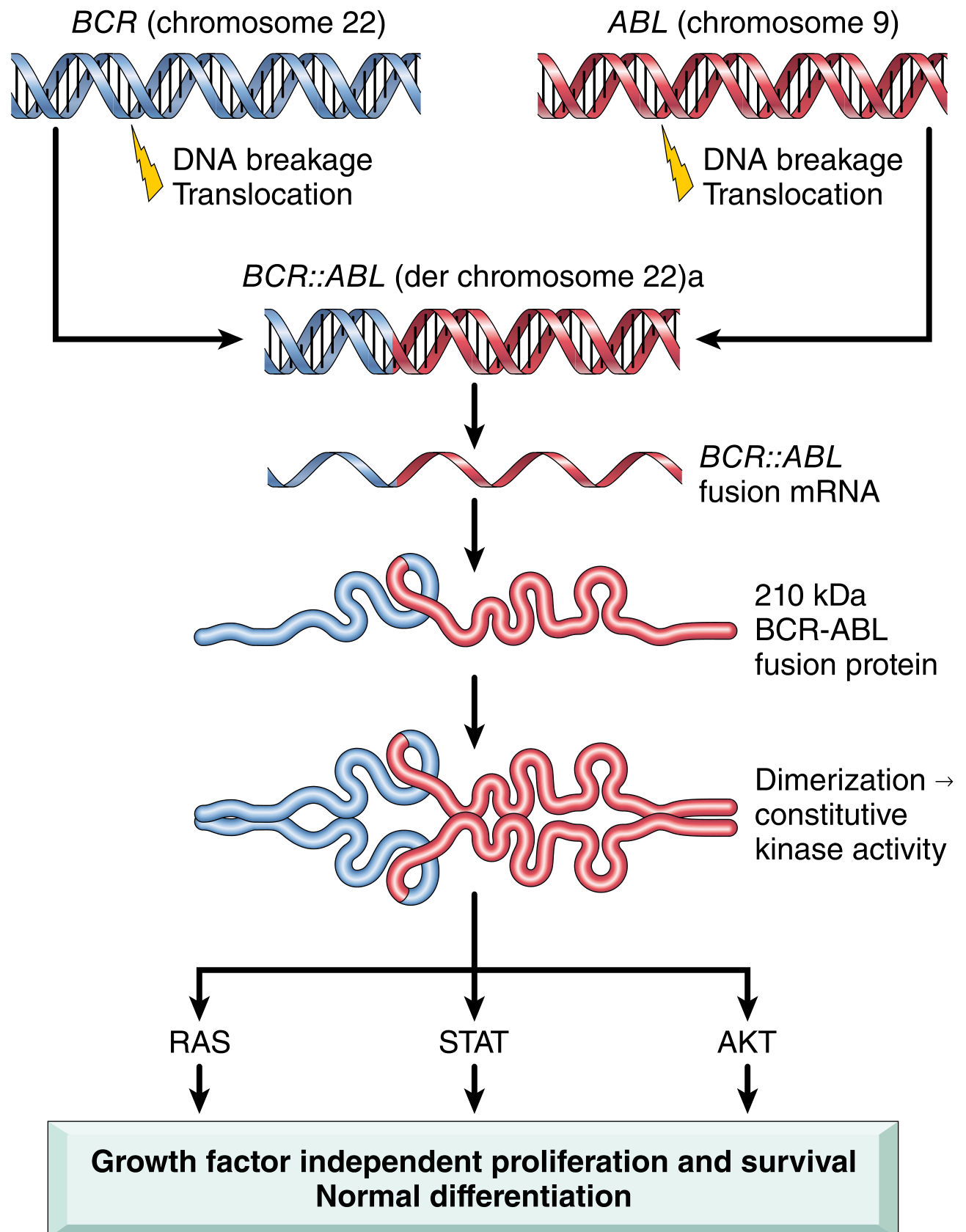
The BCR-ABL fusion gene is the molecular hallmark of CML:
- In >90% of cases, it arises from a reciprocal t(9;22)(q34;q11) translocation - the so-called Philadelphia chromosome (Ph)
- ABL (a proto-oncogene encoding a tyrosine kinase) from chromosome 9 fuses with BCR on chromosome 22
- The resulting derivative chromosome 22 carries the BCR::ABL chimeric gene
- In the remaining ~5-10% of cases, the fusion is created by cytogenetically complex or cryptic rearrangements detectable only by FISH or PCR
The BCR-ABL Oncoprotein
The fusion gene encodes a 210 kDa BCR-ABL fusion protein (p210) with a constitutively active ABL tyrosine kinase domain. The mechanism:
- BCR contains a dimerization domain that causes BCR-ABL to self-associate
- Dimerization leads to constitutive activation of the ABL kinase moiety
- The kinase phosphorylates downstream substrates, activating the RAS, JAK/STAT, and AKT pathways - the same pro-growth, pro-survival pathways normally activated by hematopoietic growth factors
Critically, BCR-ABL does not block differentiation. This explains the clinical picture: excessive production of mature, relatively normal blood cells (particularly granulocytes and platelets), rather than a block at the blast stage seen in acute leukemias.
Cell of Origin
The BCR-ABL fusion gene is found in granulocytic, erythroid, megakaryocytic, B-cell, and sometimes T-cell precursors - confirming that CML arises from a transformed pluripotent hematopoietic stem cell (HSC).
Morphology
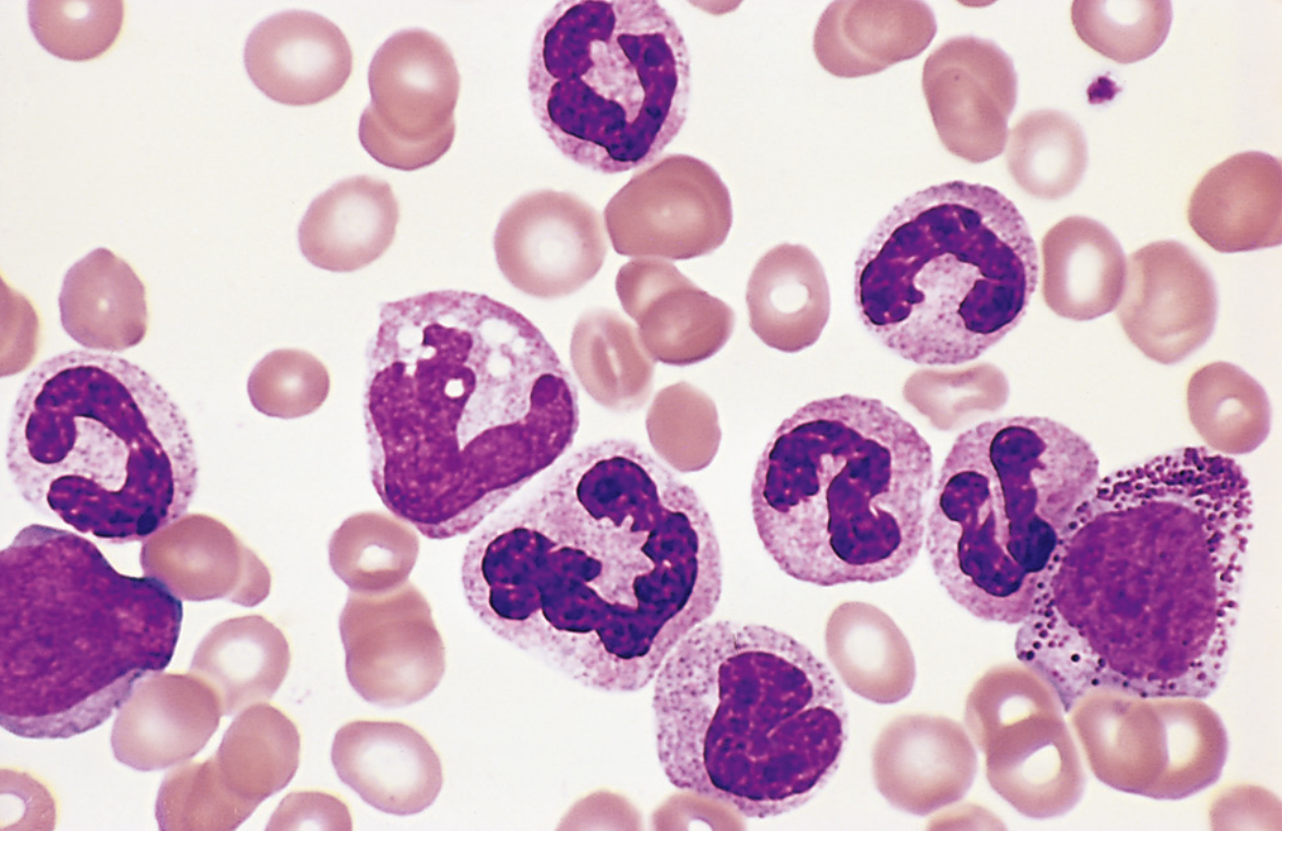
Peripheral Blood
- Leukocytosis, often exceeding 100,000 cells/µL
- Predominantly neutrophils and immature granulocytic forms (metamyelocytes, myelocytes, band forms)
- Basophilia and eosinophilia are characteristic
- Thrombocytosis (elevated platelets), often markedly so
- Blasts typically make up <10% of circulating cells in the chronic phase
Bone Marrow
- Markedly hypercellular due to massively increased maturing granulocytic precursors
- Elevated proportions of eosinophils and basophils
- Megakaryocytes are increased, often including small dysplastic forms
- Erythroid progenitors present in normal or mildly decreased numbers
- Scattered macrophages with abundant wrinkled, green-blue cytoplasm - "sea-blue histiocytes" (characteristic finding)
- Increased reticulin deposition, but overt marrow fibrosis is rare in the chronic phase
Spleen
- Often massively enlarged - up to 2630 g (normal 150-200 g)
- Red pulp resembles bone marrow due to extensive extramedullary hematopoiesis
- Frequently contains splenic infarcts of varying age due to compromised local blood supply

Clinical Features
Presentation
- Onset is insidious
- Symptoms of hypermetabolism and anaemia: fatigability, weakness, weight loss, anorexia
- A dragging sensation in the left upper abdomen due to splenomegaly is common; acute left upper quadrant pain may occur from splenic infarction
- May be an incidental finding on a routine blood count
Distinguishing CML from Leukemoid Reaction
A leukemoid reaction (dramatic granulocyte elevation due to infection, stress, or inflammation) can mimic CML. Definitive distinction is achieved by testing for the BCR-ABL fusion gene via:
- Karyotyping (looking for the Philadelphia chromosome)
- Fluorescence in situ hybridization (FISH)
- PCR-based assays
Natural History and Disease Progression
CML has three recognizable phases:
1. Chronic Phase
- Slow progression; without treatment, median survival ~3 years
- Characterised by excessive production of mature granulocytes and platelets
- BCR-ABL is the sole oncogenic driver; differentiation is preserved
2. Accelerated Phase (in ~50% of patients)
After a variable period averaging 3 years:
- Increasing anaemia and new thrombocytopenia
- Rise in basophil count
- Acquisition of additional cytogenetic abnormalities (trisomy 8, isochromosome 17q, duplication of Ph chromosome)
- Lasts 6-12 months before terminating in blast crisis
3. Blast Crisis
- Resembles acute leukemia
- In the other 50% of patients, blast crisis occurs abruptly without an accelerated phase
- 70% are myeloid blast crises; most of the remainder are lymphoid (pre-B cell) blast crises
- Lymphoid blast crisis: 85% of cases show IKZF1 mutations (Ikaros transcription factor) - the same mutations found in BCR-ABL-positive B-ALL, suggesting shared molecular origin
- Blast crisis is caused by acquisition of mutations in transcriptional regulators that block differentiation (complementing the BCR-ABL tyrosine kinase)
Treatment (Targeted Therapy)
Understanding BCR-ABL pathogenesis led directly to one of the landmark achievements in oncology:
- BCR-ABL kinase inhibitors (imatinib and successors) produce sustained hematologic remissions in >90% of patients with generally tolerable side effects
- These drugs markedly decrease BCR-ABL-positive cells in marrow and blood
- They usually do not extinguish the CML "stem cell," which persists at low levels
- However, they effectively control blood counts and substantially reduce the risk of transformation to accelerated phase and blast crisis - the greatest threats to life
- Proposed mechanism: by reducing the proliferative drive of BCR-ABL-positive progenitors, inhibitors decrease the rate at which these cells acquire additional mutations driving disease progression
Summary Table
| Feature | Detail |
|---|---|
| Mutation | BCR::ABL fusion gene (100% of cases) |
| Cytogenetics | t(9;22)(q34;q11) - Philadelphia chromosome (>90%) |
| Fusion protein | 210 kDa constitutively active tyrosine kinase |
| Cell of origin | Pluripotent HSC |
| Key blood finding | Leukocytosis >100,000/µL, basophilia, thrombocytosis |
| Marrow finding | Hypercellular; sea-blue histiocytes; increased reticulin |
| Spleen | Massively enlarged due to extramedullary haematopoiesis |
| Blast crisis | 70% myeloid; 30% lymphoid (pre-B cell) |
| Therapy | BCR-ABL kinase inhibitors (imatinib); >90% remission rate |
Sources: Robbins, Cotran & Kumar - Pathologic Basis of Disease (10th ed.); Robbins & Kumar - Basic Pathology

image.png
From harsh mohon
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
I now have all the information needed for a thorough, exam-style answer to both questions. Here it is:
4. Differences — Haemoglobinopathy, Thalassaemia & Sickle Cell Anaemia
(Robbins Cotran & Kumar — Pathologic Basis of Disease)
a. Thalassaemia vs. Haemoglobinopathy (2016, 2015)
These two categories of inherited red cell disorders are often confused because they overlap, but they are fundamentally distinct at the molecular level.
| Feature | Thalassaemia | Haemoglobinopathy |
|---|---|---|
| Basic defect | Quantitative - decreased synthesis of structurally normal globin chains | Qualitative - synthesis of a structurally abnormal (mutant) globin chain |
| Mechanism | Mutations reduce or abolish globin chain production (β⁰ or β⁺) | Point mutations alter the amino acid sequence of a globin chain |
| Globin protein | Normal in structure, but produced in reduced amounts | Abnormal amino acid sequence - e.g., Glu→Val at position 6 of β-globin (HbS) |
| Prototype | β-thalassaemia, α-thalassaemia | Sickle cell disease (HbS), HbC disease, HbE disease |
| Molecular lesion | Splicing mutations, promoter mutations, chain terminator mutations (nonsense/frameshift) | Missense mutation in the globin gene |
| Pathogenic basis | Imbalanced globin chain synthesis → unpaired chains precipitate → membrane damage, ineffective erythropoiesis | Physicochemical abnormality of the mutant Hb (e.g., HbS polymerises under hypoxia) |
| Type of anaemia | Hypochromic, microcytic; largely due to ineffective erythropoiesis + haemolysis | Normocytic haemolytic anaemia (in SCA) |
| HbA level | Reduced (in β-thal) | Normal amount of Hb produced, but it is the wrong type |
| HbF | Markedly elevated in β-thal major (compensatory) | May be elevated if hereditary persistence of HbF co-exists |
| Iron status | Iron overload (due to increased absorption via erythroferrone + transfusions) | Iron overload less prominent unless transfusion-dependent |
| Bone changes | Prominent - "crew-cut" skull on X-ray from marrow hyperplasia | Seen in SCA but less extreme |
| Geographic prevalence | Mediterranean, Middle East, India, Southeast Asia | West Africa, African Americans, Middle East |
| Overlap | Thalassaemia is technically also a haemoglobinopathy in the broad sense, but the classification separates them by qualitative vs. quantitative defect |
Key Concept
Thalassaemias = too little of a normal globin chain. Haemoglobinopathies = normal amount of an abnormal globin chain.
Notably, both categories can coexist: a patient may inherit β-thalassaemia on one chromosome and HbS on the other (HbS/β-thal), producing a sickling disorder of variable severity.
b. Thalassaemia vs. Sickle Cell Anaemia (SCA) (2021)
Both are autosomal recessive disorders of haemoglobin - but they differ in almost every pathological detail:
| Feature | Thalassaemia (β-thal major) | Sickle Cell Anaemia |
|---|---|---|
| Category | Quantitative haemoglobinopathy (↓ globin synthesis) | Qualitative haemoglobinopathy (structurally abnormal Hb) |
| Mutation | Multiple mutations in β-globin gene (splicing, promoter, chain terminator) | Single missense point mutation: Glu→Val at codon 6 of β-globin gene |
| Abnormal protein | No structural abnormality in globin; simply ↓ β-chain production | HbS (α₂β²ˢ) - valine substitution gives hydrophobic patch on deoxyHbS |
| Pathogenic mechanism | Unpaired α-chains precipitate → membrane damage → ineffective erythropoiesis + haemolysis | HbS polymerises when deoxygenated → sickling → haemolysis + microvascular occlusion |
| Primary problem | Ineffective erythropoiesis (most precursors die in marrow) + haemolysis | Haemolysis + vascular occlusion |
| Type of haemolysis | Predominantly intravascular destruction of precursors + extravascular haemolysis | Primarily extravascular (spleen, liver); also intravascular due to fragile sickled cells |
| Blood smear | Hypochromic, microcytic cells; target cells, poikilocytes, anisocytes, nucleated RBCs, basophilic stippling | Sickle cells (irreversibly sickled cells), target cells, Howell-Jolly bodies, reticulocytosis |
| HbA | Absent (β⁰/β⁰) or markedly reduced | Absent (replaced entirely by HbS in homozygotes) |
| HbF | Markedly elevated (compensatory) | Normally low after 6 months; if elevated, disease is milder |
| HbA₂ | Elevated (β-thal minor/trait) | Normal |
| Anaemia severity | Severe (Hb 3-6 g/dL in untransfused β-thal major) | Moderate (haematocrit 18%-30%) |
| Spleen | Enlarged (extramedullary haematopoiesis) - splenomegaly throughout life | Enlarged in infancy (sickled cells trapped) → progressive infarction → autosplenectomy by adolescence (tiny fibrotic remnant) |
| Bone marrow | Massive erythroid hyperplasia, bony cortex erosion | Erythroid hyperplasia; "crew-cut" skull on X-ray |
| Skeletal changes | Prominent: frontal bossing, maxillary hypertrophy, "crew-cut" skull | Present but less pronounced |
| Iron overload | Major complication - from gut absorption (erythroferrone ↑ hepcidin suppression) + transfusions → secondary haemochromatosis, cardiac/hepatic injury | Less prominent unless heavily transfused |
| Vascular occlusion | NOT a feature | Cardinal feature - bone pain crises, stroke, acute chest syndrome, renal infarcts, leg ulcers, retinal infarcts |
| Painful crises | Absent | Present - the hallmark; triggered by hypoxia, dehydration, infection, cold |
| Infections | Increased (due to splenomegaly-related immune dysfunction; also transfusion risks) | Greatly increased due to autosplenectomy; especially encapsulated organisms (Streptococcus pneumoniae, Haemophilus influenzae) |
| Gallstones | Pigment gallstones (from chronic haemolysis) | Pigment gallstones (from chronic haemolysis) - both share this |
| Malaria protection | Heterozygous carriers (thalassaemia trait) have some protection | Heterozygous HbS carriers (sickle cell trait) protected against P. falciparum malaria |
| Treatment | Transfusions + iron chelation; hydroxyurea; stem cell transplant; gene therapy | Hydroxyurea (↑ HbF); transfusions for crises; stem cell transplant; analgesics |
| Prognosis | Untreated β-thal major: death in 1st or 2nd decade from iron overload | Untreated: median survival ~40-50 years; modern treatment improves this |
Shared Features (important to note)
- Both are autosomal recessive
- Both cause haemolytic anaemia with erythroid hyperplasia
- Both produce pigment gallstones and splenomegaly (at least initially)
- Both are most common in populations where falciparum malaria is/was endemic (heterozygote advantage)
- Both can cause extramedullary haematopoiesis in severe disease
Sources: Robbins, Cotran & Kumar - Pathologic Basis of Disease (Robbins Cotran Pathology); Robbins & Kumar - Basic Pathology
This is a shared conversation. Sign in to Orris to start your own chat.