possible etiology of lead posioning in 3 yr old child , workup on verbal delay to ruke out ala to protoporpyrin conversion
Reading File
Checking Date & Time
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Searching PubMed
lead poisoning children etiology sources
Reading File
I now have comprehensive information to provide a thorough answer. Let me compile it.
Lead Poisoning in a 3-Year-Old: Etiology & Workup
1. Etiology / Sources of Lead Exposure in Young Children
Young children (especially ages 1–5) are disproportionately vulnerable because of hand-to-mouth behavior, higher GI absorption (~50% vs ~10% in adults), and a developing nervous system. Common sources:
Environmental / Household
| Source | Mechanism |
|---|---|
| Lead-based paint (most common in US) | Chipping/peeling paint in pre-1978 homes; pica behavior (eating paint chips) |
| Lead-contaminated dust & soil | Paint dust from renovation, near old buildings or highways |
| Lead pipes / plumbing solder | Leaching into drinking water (especially first-flush tap water) |
| Contaminated soil | Near former industrial sites, smelters, or high-traffic roads (leaded gasoline residue) |
Ingested / Product-Related
| Source | Mechanism |
|---|---|
| Traditional/herbal remedies | Ayurvedic medicines, azarcon (Mexican folk remedy), greta, pay-loo-ah — common cause globally (PMID 36322973) |
| Imported toys, jewelry, trinkets | High lead content in painted/coated items from some countries |
| Glazed pottery / ceramics | Lead-glazed cookware or dishware leaching into food/beverages |
| Cosmetics (kohl, surma) | Eye cosmetics used in South Asian/Middle Eastern households |
| Contaminated food/spices | Imported spices, candies (tamarind candies notably implicated) |
Occupational / Secondary (Take-home lead)
- Parent working in battery recycling, radiator repair, auto mechanics, construction/demolition
- Lead brought home on clothing, shoes, or hands
Other
- Retained foreign body (e.g., bullet fragment, fishing sinker ingested or embedded)
- Burning painted wood or batteries for fuel (low-income settings)
— Robbins Pathologic Basis of Disease: "For most of the twentieth century the major sources of lead in the environment were lead-containing house paints and gasoline."
— Henry's Clinical Diagnosis: "Lead poisoning is seen in children in large cities who consume lead in the form of paint (pica)."
2. Pathophysiology: ALA → Protoporphyrin Conversion (The Key Enzymatic Block)
Understanding the biochemical defect is essential to interpreting the workup:
Normal Heme Biosynthesis Pathway
ALA synthetase
Glycine + Succinyl-CoA ──────────────► δ-ALA (delta-aminolevulinic acid)
↓
δ-ALA dehydratase (ALAD) ← [BLOCKED BY LEAD]
↓
Porphobilinogen
↓
(multiple steps) → Coproporphyrinogen III
↓
Protoporphyrin IX
↓
Ferrochelatase ← [BLOCKED BY LEAD]
(Fe²⁺ + Protoporphyrin IX → Heme)
Lead blocks two key enzymes:
- δ-ALA dehydratase (ALAD) — prevents ALA → porphobilinogen conversion → ALA accumulates in urine
- Ferrochelatase — prevents iron insertion into protoporphyrin IX → protoporphyrin accumulates, zinc substitutes → zinc protoporphyrin (ZPP) forms instead of heme
Additional enzyme inhibited: Coproporphyrinogen decarboxylase — causing coproporphyrinuria
Lead also inhibits pyrimidine-5'-nucleotidase → RNA clumping → basophilic stippling of RBCs
— Henry's Clinical Diagnosis: "Lead blocks the action of β-ALA synthetase, δ-ALA dehydratase (ALAD), coproporphyrinogen decarboxylase, and ferrochelatase, producing anemia."
— Robbins Basic Pathology: "Zinc-protoporphyrin (ZPP) is formed instead of heme, leading to decreased iron incorporation into heme and subsequent anemia."
3. Workup for a 3-Year-Old with Verbal Delay (to Exclude Lead Toxicity as Cause)
First-Line: Blood Lead Level (BLL)
- Whole blood lead measured by atomic absorption spectroscopy or ICP/MS
- Reference & action thresholds (CDC/AAP):
- < 3.5 µg/dL — current CDC reference value (updated 2021; previously 5 µg/dL)
- ≥ 3.5 µg/dL — requires follow-up
- ≥ 45 µg/dL — chelation therapy indicated
- ≥ 70 µg/dL — medical emergency
- BLL reflects recent exposure; short half-life (~35 days in blood)
Heme Pathway Markers (confirming ALA → Protoporphyrin block)
| Test | What it Detects | Clinical Notes |
|---|---|---|
| Zinc Protoporphyrin (ZPP) / Erythrocyte Zinc Protoporphyrin | Zinc substitution when ferrochelatase is blocked | Simple fluorometric assay; elevated in frank toxicity but not sensitive at low BLL (< 25 µg/dL) |
| Free Erythrocyte Protoporphyrin (FEP) | Accumulated protoporphyrin IX from blocked ferrochelatase | Elevated when BLL > 25–35 µg/dL; not useful for current low-level screening |
| Urine δ-ALA | ALA accumulation from ALAD block | Elevated with inorganic lead poisoning; useful marker |
| ALAD activity in RBCs | Direct measure of enzyme inhibition | Most sensitive for organolead poisoning |
| Urine coproporphyrin | Coproporphyrinogen decarboxylase block | Elevated; less commonly tested |
— Henry's Clinical Diagnosis: "Increased amounts of ALA in urine, decreased ALAD activity in red blood cells, increased amounts of free erythrocyte protoporphyrin, and elevated amounts of zinc protoporphyrin are found with inorganic lead poisoning. The assay for zinc protoporphyrin is a particularly simple fluorometric one that is widely used and is an excellent screening test for frank lead toxicity. However, the test is not sensitive to the low blood lead levels now regarded as harmful."
— Robbins Basic Pathology: "Elevated blood lead, red cell free protoporphyrin, or zinc-protoporphyrin levels are required for definitive diagnosis."
Complete Blood Count (CBC)
- Microcytic, hypochromic anemia
- Basophilic stippling of RBCs — classic, but not pathognomonic
- Hemolysis markers if indicated
Imaging
- Long bone X-rays (wrist/knee) — "lead lines" at growth plates (radiopaque bands at physis); indicates chronic exposure
- Abdominal X-ray — if pica suspected, to identify ingested paint chips or foreign bodies
Renal Function
- Urinalysis: albuminuria, hematuria (proximal tubular damage)
- BMP/CMP: creatinine
Neurodevelopmental Assessment (directly relevant to verbal delay)
- Formal developmental evaluation (Bayley Scales, ADOS, speech-language assessment)
- Lead neurotoxicity causes: reduced IQ, learning disabilities, retarded psychomotor development, verbal/language delay; these CNS effects in children are usually irreversible
Environmental Assessment
- Detailed environmental history: housing age, renovation, water source, parental occupation, dietary history, folk medicine use, pica behavior
- Home lead inspection / EPA-certified lead assessment if indicated
Summary Table: Heme Pathway Workup
| Level | Test | Pathologic Basis |
|---|---|---|
| Substrate accumulation | Urine δ-ALA ↑ | ALAD blocked → ALA can't convert to porphobilinogen |
| Enzyme activity | RBC ALAD activity ↓ | Direct inhibition by lead |
| End-product | FEP or ZPP ↑ | Ferrochelatase blocked → Zn²⁺ inserts instead of Fe²⁺ |
| Definitive | Whole blood lead level | Gold standard for recent exposure |
| Cumulative burden | Bone lead (XRF) | Half-life in bone ~32 years; research setting |
Note on verbal delay: At blood lead levels as low as 10 µg/dL (now considered harmful), cognitive and language delays occur without overt encephalopathy. The ZPP/FEP tests may be normal at these levels — BLL remains the only reliable screen for low-level neurotoxic exposure.
— Robbins Pathologic Basis: "Effects of chronic lead exposure in children may be subtle, producing mild dysfunction, including reduced IQ, learning disabilities, and delayed psychomotor development."
what is jak 2 kinase
Reading File
Checking Date & Time
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Loading Image
Reading File
Excellent — I now have all the material needed for a comprehensive answer. Here it is:
JAK2 Kinase
What Is JAK2?
JAK2 (Janus Kinase 2) is a non-receptor cytoplasmic protein tyrosine kinase — it sits attached to the inner surface of the cell membrane rather than being a transmembrane receptor itself. It belongs to the Janus kinase (JAK) family, named after the two-faced Roman god because each JAK has two kinase-like domains: one active (JH1) and one pseudokinase/autoinhibitory (JH2).
The JAK-STAT Signaling Pathway (Normal Function)
JAK2 is the central mediator of cytokine receptor signaling, particularly for hematopoietic growth factors. The pathway works in five steps:
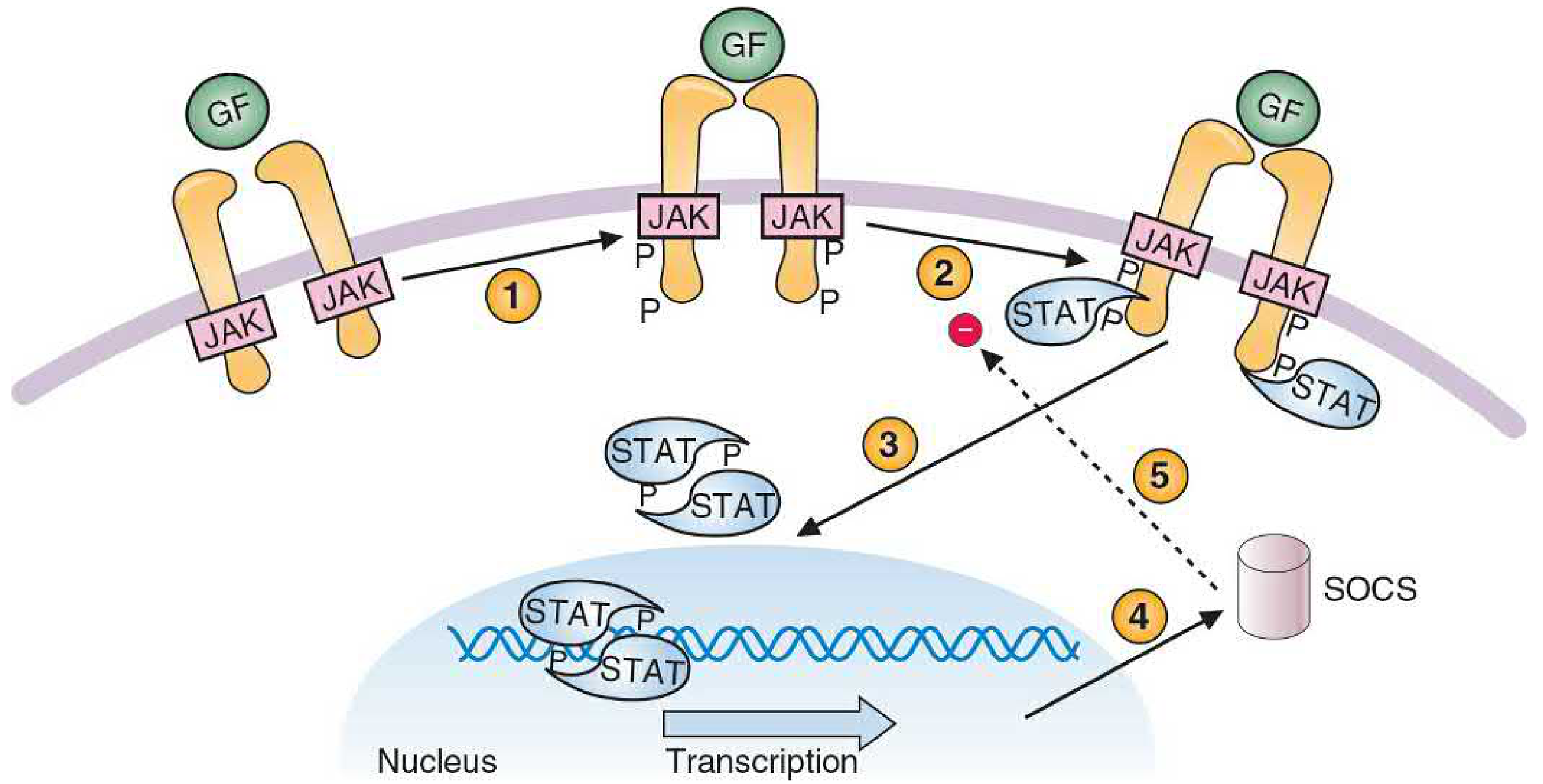
JAK-STAT pathway: (1) Growth factor binds → receptor dimerizes → JAKs phosphorylate each other. (2) Activated JAKs phosphorylate the receptor, creating docking sites for STAT proteins. (3) STATs are phosphorylated, dimerize, and translocate to the nucleus. (4) STAT dimers activate target gene transcription. (5) SOCS proteins are induced as negative feedback to inhibit JAKs. — Basic Medical Biochemistry, 6e
Step-by-step:
- Cytokine/growth factor (e.g., erythropoietin, thrombopoietin, IL-2, IL-3) binds its receptor
- Receptor dimerizes → JAK2 molecules (constitutively associated with the receptor) transphosphorylate each other (activation)
- Activated JAK2 phosphorylates the cytokine receptor → creates docking sites for STAT proteins (Signal Transducers and Activators of Transcription)
- JAK2 phosphorylates STATs → they dimerize and translocate to the nucleus → activate target genes (proliferation, differentiation, anti-apoptosis)
- JAK2 also activates the Ras/Raf/MAP kinase and PI3K/AKT pathways in parallel
- SOCS (Suppressor of Cytokine Signaling) proteins are induced as negative feedback to shut JAK2 off
Key cytokines signaling through JAK2:
- Erythropoietin (EPO) → red cell production
- Thrombopoietin (TPO/MPL) → platelet production
- G-CSF, GM-CSF → granulopoiesis
- Growth hormone, prolactin, leptin
JAK2 Mutations in Disease: The V617F Mutation
The clinically critical mutation is JAK2 V617F (c.1849G>T, p.Val617Phe):
- Located in the JH2 pseudokinase (autoinhibitory) domain
- Normally JH2 suppresses JH1 kinase activity; the V617F substitution disrupts this autoinhibition → constitutive (ligand-independent) kinase activation
- Results in continuous signaling through JAK-STAT even without growth factor binding
- Hematopoietic progenitors become EPO-independent — they proliferate without needing erythropoietin
— Tietz Textbook of Laboratory Medicine: "The JAK2 p.Val617Phe mutation occurs in the autoinhibitory pseudokinase (JH2) domain of JAK2 and imparts dysregulated kinase activity... polycythemia vera progenitor cells are abnormally EPO independent."
Disease Associations
| Disease | JAK2 V617F Frequency | Effect |
|---|---|---|
| Polycythemia vera (PV) | >95% | Constitutive JAK2 activation → uncontrolled erythropoiesis |
| Essential thrombocythemia (ET) | 50–60% | Thrombocytosis |
| Primary myelofibrosis (PMF) | 50–60% | Marrow fibrosis, splenomegaly |
| Myeloid/lymphoid neoplasms with eosinophilia | Rare fusion variants | Constitutive activation |
In ET and PMF where JAK2 V617F is absent, alternative mutations activate the same pathway: CALR mutations (25–35%) or MPL mutations (5–10%).
— Robbins Pathologic Basis of Disease: Mutations in JAK2, CALR, and MPL all result in constitutive tyrosine kinase activation in myeloproliferative neoplasms.
Homozygosity: Many MPN patients have homozygous JAK2 V617F due to loss of heterozygosity (LOH) via mitotic recombination at chromosome 9p24 (the JAK2 locus) — correlates with more severe disease phenotype.
Clinical Use: JAK2 V617F Testing
- Diagnostic test for suspected myeloproliferative neoplasms (MPNs)
- Distinguishes neoplastic from reactive causes of erythrocytosis/thrombocytosis
- Detected by: allele-specific PCR, pyrosequencing, next-generation sequencing (NGS) myeloid panels
- A positive result is a WHO diagnostic criterion for PV, ET, and PMF
JAK2 Inhibitors (Therapeutic Targeting)
| Drug | Targets | Approved Indication |
|---|---|---|
| Ruxolitinib | JAK1 + JAK2 | Myelofibrosis, PV (hydroxyurea-resistant) |
| Fedratinib | JAK2 (+ FLT3) | Myelofibrosis |
| Pacritinib | JAK2 + IRAK1 | Myelofibrosis with thrombocytopenia |
| Momelotinib | JAK1 + JAK2 + ACVR1 | Myelofibrosis (also reduces anemia via hepcidin suppression) |
— Tietz: "Ruxolitinib treatment reduced patient spleen size and improved the overall burden of symptoms... JAK2 inhibitor therapy markedly improves quality of life of MPN patients but, in contrast to imatinib in CML, may not improve overall survival."
Summary
| Feature | Detail |
|---|---|
| Type | Non-receptor cytoplasmic tyrosine kinase |
| Location | Chromosome 9p24 |
| Normal function | Signal transduction downstream of cytokine receptors (EPO, TPO, etc.) via JAK-STAT pathway |
| Oncogenic mechanism | V617F point mutation → loss of JH2 autoinhibition → constitutive activation |
| Diseases | Polycythemia vera (>95%), ET (50–60%), primary myelofibrosis (50–60%) |
| Inhibitors | Ruxolitinib, fedratinib, pacritinib, momelotinib |
| Negative regulators | SOCS proteins |
what is aplastic crisis
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Loading Image
Comprehensive answer ready:
Aplastic Crisis
Definition
Aplastic crisis is an abrupt, temporary cessation of red blood cell production (erythropoiesis) in the bone marrow. It is not true aplastic anemia (which involves all cell lines from the start) — it is specifically a sudden arrest of the erythroid lineage that causes a rapid, severe drop in hemoglobin, most dangerous in patients who already have shortened RBC survival.
Cause: Parvovirus B19
The etiology in virtually all cases is Parvovirus B19 (a small, non-enveloped, single-stranded DNA virus).
— Goldman-Cecil Medicine: "The anemic crises associated with low or absent reticulocytes in hereditary spherocytosis and sickle cell disease are virtually always secondary to B19 parvovirus infection."
— Jawetz Medical Microbiology: "Parvovirus B19 is the cause of transient aplastic crisis that may complicate chronic hemolytic anemia, such as in patients with sickle cell disease, thalassemias, and acquired hemolytic anemias."
Mechanism: Why B19 Stops Erythropoiesis
The receptor: Globoside (Blood Group P antigen)
- B19 binds globoside (P antigen), which is abundantly expressed on erythroid progenitors, erythroblasts, megakaryocytes, and endothelial cells
- The virus enters the cell via P antigen → replicates in the nucleus of mitotically active immature erythroid cells → causes cell lysis and arrest of erythroid development
- CFU-E (colony-forming unit – erythroid) cells are the primary target
Why normal people don't notice
- Normal RBC lifespan = 120 days → a 7–10 day interruption in production barely changes hemoglobin
- Erythropoiesis resumes once the immune system produces neutralizing antibodies
Why it is catastrophic in hemolytic anemia
- In sickle cell disease, thalassemia, hereditary spherocytosis, etc., RBC lifespan is drastically shortened (10–30 days)
- The marrow is already working at maximum compensatory output just to maintain baseline hemoglobin
- Even a brief arrest of production causes precipitous, life-threatening anemia
— Sherris Medical Microbiology: "The clinical consequences of this effect on erythrocytes are generally trivial, unless patients are already compromised by a chronic hemolytic process, such as sickle cell disease or thalassemia, in which maximal erythropoiesis is continually needed to counterbalance increased destruction of circulating erythrocytes."
Clinical Features
| Feature | Detail |
|---|---|
| Preceding illness | Fever, malaise ~7–10 days before hematologic crisis (viremic phase) |
| Rash | Usually absent (rash of fifth disease appears only after viremia clears, when crisis is over) |
| Hemoglobin | Rapid, severe fall |
| Reticulocytes | Markedly reduced or absent (reticulocytopenia — key finding) |
| WBC / platelets | May also fall, especially with functioning spleen |
| Duration | Self-limited — erythroid recovery in 1–2 weeks once antibodies form |
| Complications | Severe anemia → heart failure, cerebrovascular accident; can be fatal |
— Goldman-Cecil: "Although it is self-limited, aplastic crisis often requires transfusion and can lead to severe, occasionally fatal anemia that precipitates heart failure and cerebrovascular accidents."
Bone Marrow Hallmark
Giant pronormoblasts — the pathognomonic cytopathic effect of B19 infection:
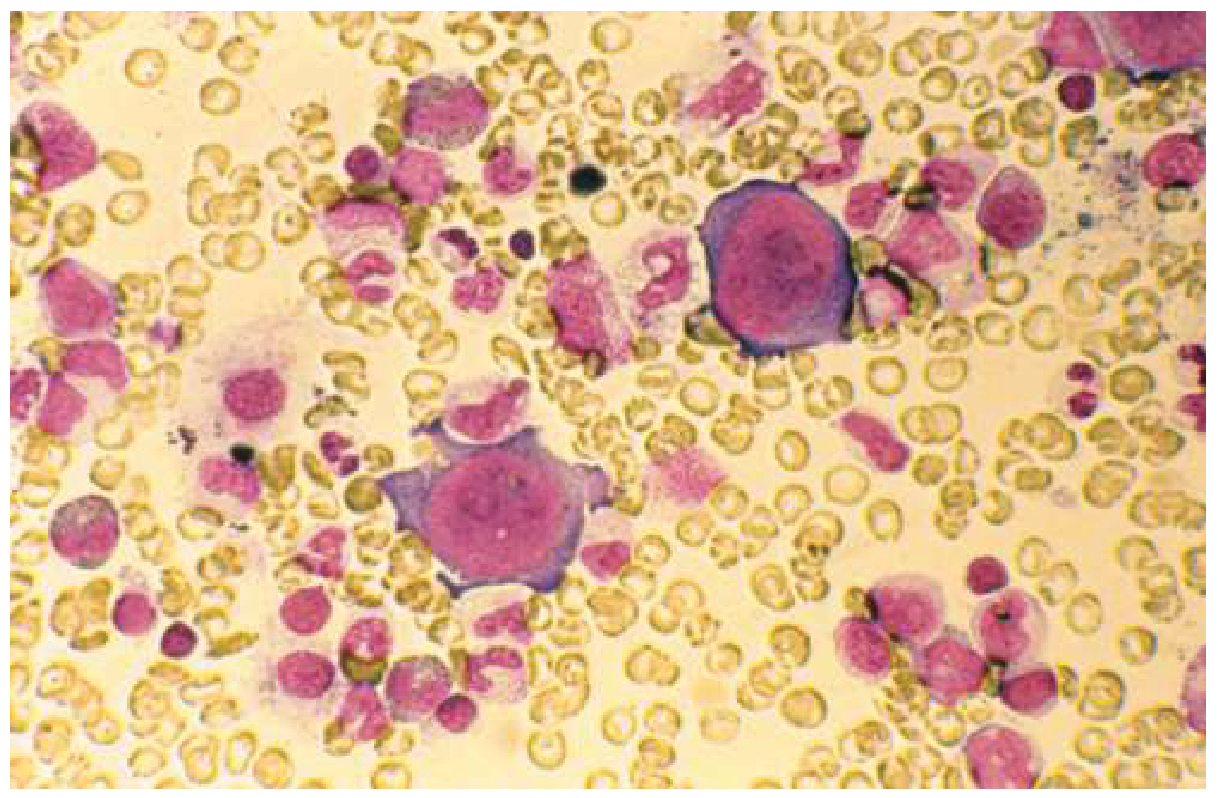
Bone marrow aspirate in B19 parvovirus aplastic crisis: Large giant pronormoblasts (infected cells) with near-complete absence of maturing erythroid precursors. — Goldman-Cecil Medicine
- Mature erythroid precursors are absent
- Scattered giant pronormoblasts are the cytopathic effect of the virus
- Granulocytic and megakaryocytic lines are relatively preserved (distinguishing from aplastic anemia)
At-Risk Conditions
Any condition causing chronic hemolysis or increased RBC demand:
| Condition | Risk |
|---|---|
| Sickle cell disease | High — most common presentation |
| Hereditary spherocytosis | High |
| Thalassemias | High |
| G6PD deficiency | Moderate |
| Autoimmune hemolytic anemia | Moderate |
| Post-bone marrow transplant | Present |
| Immunocompromised patients | Risk of persistent pure red cell aplasia (not just transient) |
Persistent Infection vs. Transient Crisis
| Transient Aplastic Crisis | Persistent B19 / Pure Red Cell Aplasia | |
|---|---|---|
| Host | Immunocompetent with chronic hemolysis | Immunocompromised (AIDS, chemotherapy, transplant) |
| Duration | 1–2 weeks, self-limited | Weeks–months, ongoing |
| Resolution | Antibody formation clears virus | Cannot make neutralizing antibodies → virus persists |
| Treatment | Supportive ± transfusion | IV immunoglobulin (IVIG) to supply exogenous antibodies |
Management
- Supportive care: rest, monitoring hemoglobin
- RBC transfusion: when anemia is symptomatic or hemoglobin critically low
- Infection control: B19 is contagious via respiratory droplets → isolate from immunocompromised and pregnant contacts
- IVIG: for immunocompromised patients with persistent pure red cell aplasia
- No specific antiviral therapy available
Key Distinguishing Point
Aplastic crisis is not aplastic anemia:
| Aplastic Crisis | Aplastic Anemia | |
|---|---|---|
| Cause | Parvovirus B19 | Autoimmune, drugs, radiation |
| Cell lines affected | Erythroid only (primarily) | All cell lines (pancytopenia) |
| Duration | Transient (1–2 weeks) | Persistent if untreated |
| Reticulocytes | Absent | Absent |
| Bone marrow | Giant pronormoblasts, erythroid arrest | Hypocellular, fatty replacement |
— Henry's Clinical Diagnosis: "The aplastic crises resulting from parvovirus infection are transient, with erythroid marrow recovery in 1 to 2 weeks after onset."
This is a shared conversation. Sign in to Orris to start your own chat.