explain acute inflammation to medical student
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Loading Image
I now have comprehensive content from Robbins. Let me compile the full teaching explanation.
Acute Inflammation
Based on Robbins & Kumar Basic Pathology, 11e
What Is Acute Inflammation?
Acute inflammation is the rapid, stereotyped response of vascularized tissue to infection or tissue damage. Its purpose is to deliver leukocytes and plasma proteins to wherever they are needed, eliminate the offending agent, and set the stage for repair. It develops within minutes to hours and lasts days.
The classic clinical signs - described by Celsus and Virchow - map directly to the underlying mechanisms:
| Sign | Latin | Mechanism |
|---|---|---|
| Redness (erythema) | Rubor | Vasodilation |
| Heat | Calor | Vasodilation + increased blood flow |
| Swelling | Tumor | Edema from increased vascular permeability |
| Pain | Dolor | Prostaglandins, bradykinin on nerve endings |
| Loss of function | Functio laesa | Combination of above |
The Three Core Components
Acute inflammation has three major components, all occurring primarily in postcapillary venules:
- Dilation of small vessels
- Increased vascular permeability
- Emigration of leukocytes from the circulation
Component 1: Vascular Reactions
Vasodilation
- One of the earliest events
- Caused primarily by histamine (released from mast cells) and later prostaglandins and nitric oxide
- Produces redness and warmth at the site
- Slows blood flow - setting the stage for leukocyte margination
Increased Vascular Permeability (Leakage)
- Histamine, bradykinin, leukotrienes C4/D4/E4 cause endothelial cells to contract, creating gaps between them
- This happens within 15-30 minutes and is usually short-lived
- In burns, direct endothelial injury causes immediate and sustained leakage
This leakage produces an exudate - a protein-rich fluid that escapes into the interstitium. This is distinct from a transudate (seen in heart failure, liver disease), which is a low-protein ultrafiltrate produced without inflammation:
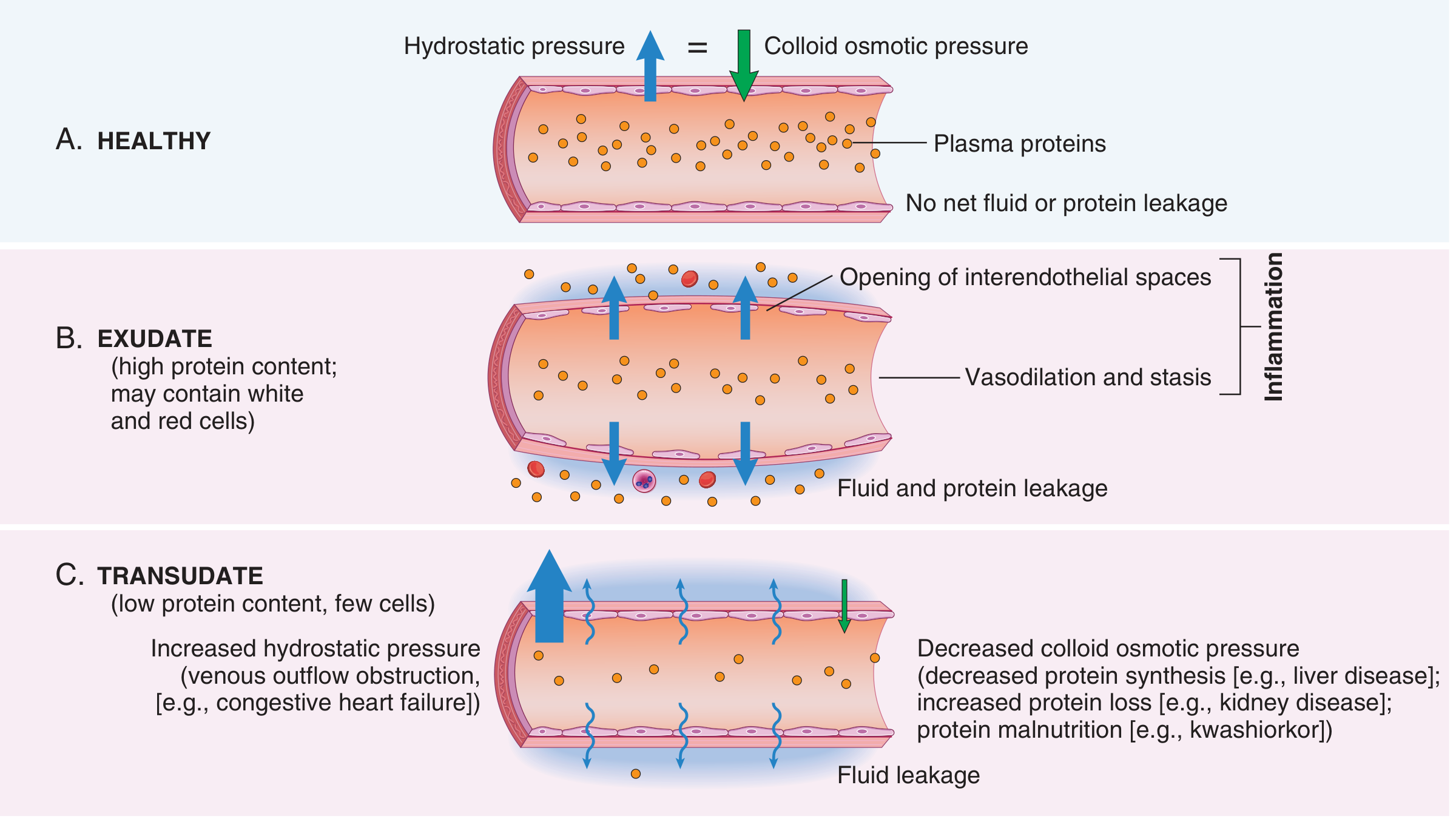
The accumulated interstitial fluid is edema. When the exudate is full of neutrophils, it is called pus.
Component 2: Leukocyte Recruitment
This is the most important component - getting neutrophils to the site. It happens in a precise, stepwise sequence:
Step-by-Step Process
1. Margination
- As blood flow slows (stasis), red cells migrate centrally
- Leukocytes are pushed peripherally toward the vessel wall
2. Rolling
- Leukocytes loosely attach to activated endothelium and tumble along it
- Mediated by selectins: E-selectin and P-selectin on endothelium bind carbohydrate ligands on leukocytes; L-selectin on leukocytes binds endothelial ligands
- P-selectin is stored in Weibel-Palade bodies and moves to the surface within minutes of histamine/thrombin stimulation
3. Firm Adhesion (Arrest)
- Chemokines (e.g., CXCL8/IL-8, CCL2) displayed on the endothelial surface activate leukocyte integrins, converting them from low-affinity to high-affinity state
- Activated integrins (LFA-1, Mac-1) bind ICAM-1 on endothelium - producing stable adhesion
4. Transmigration (Diapedesis)
- Leukocytes squeeze between endothelial cells
- Mediated by PECAM-1 (CD31), expressed on both leukocytes and endothelial junctions
5. Chemotaxis
- Leukocytes migrate through extracellular matrix toward the source of injury along a chemical gradient
- Key chemoattractants: C5a (complement), LTB4 (leukotriene B4), IL-8/CXCL8, and bacterial products (fMLP)
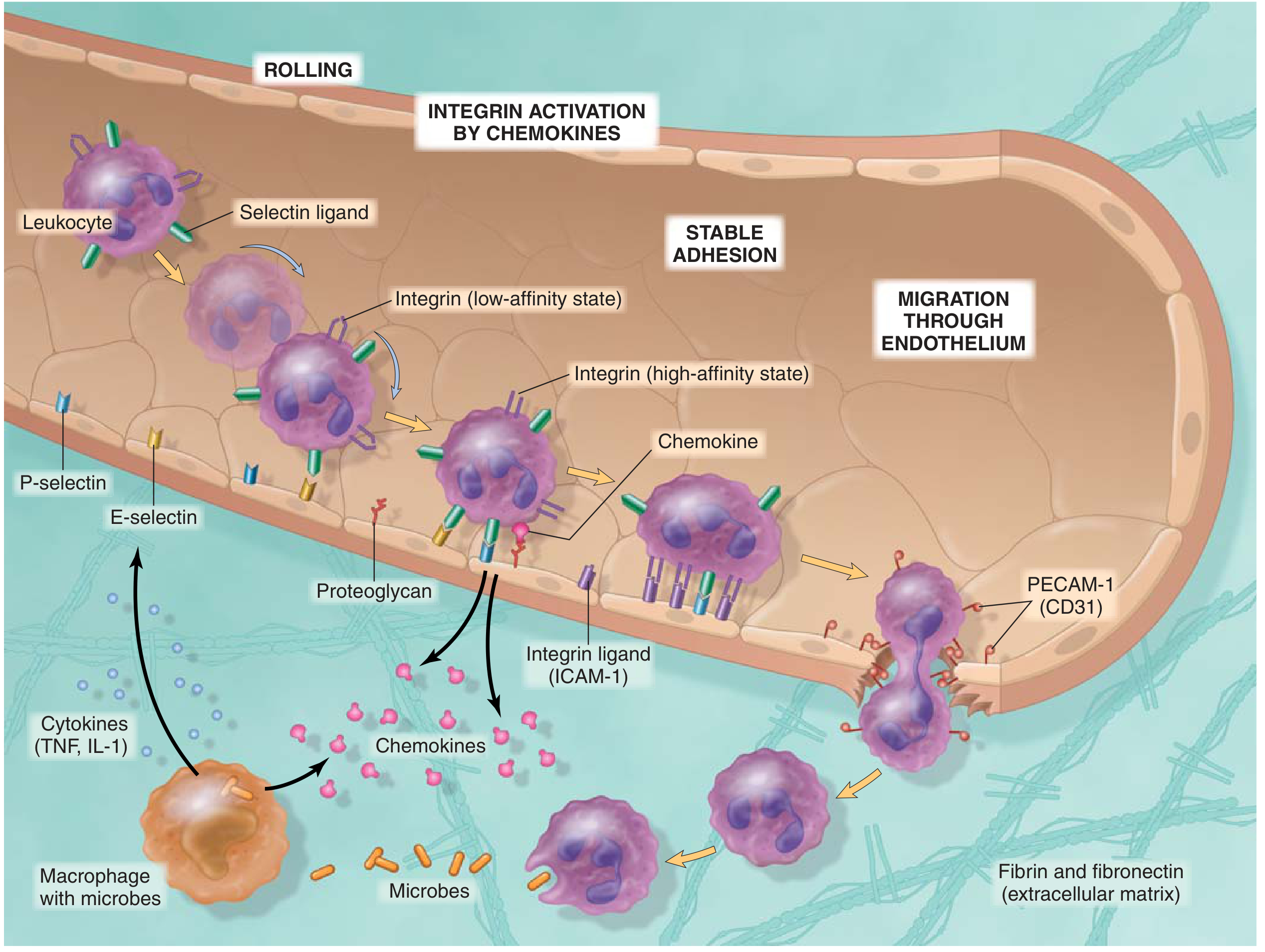
Clinical relevance: Leukocyte adhesion deficiency (LAD) is a genetic defect in integrin expression (CD18). Affected patients have recurrent bacterial infections, no pus formation, and markedly elevated blood neutrophil counts.
Phagocytosis and Killing
Once at the site, neutrophils and macrophages:
- Recognize microbes (aided by opsonins - IgG, C3b)
- Engulf them into phagosomes
- Kill them via:
- Reactive oxygen species (ROS) generated by NADPH oxidase ("oxidative burst")
- Nitric oxide (NO) via iNOS
- Lysosomal enzymes (elastase, myeloperoxidase, defensins)
Neutrophil Extracellular Traps (NETs): Activated neutrophils can eject their chromatin as fibrillar networks that trap and kill extracellular microbes - at the cost of their own death (NETosis).
Component 3: Mediators of Inflammation
Mediators orchestrate all the above events. They are produced locally at the site and are short-lived. The main ones to know:
| Reaction | Key Mediators |
|---|---|
| Vasodilation | Histamine, prostaglandins (PGI2), NO |
| Increased permeability | Histamine, C3a/C5a, leukotrienes C4/D4/E4 |
| Chemotaxis/leukocyte recruitment | C5a, LTB4, IL-8, TNF, IL-1 |
| Fever | IL-1, TNF, IL-6, prostaglandins (PGE2) |
| Pain | Prostaglandins, bradykinin, substance P |
| Tissue damage | Lysosomal enzymes, ROS |
The Major Mediator Families
Vasoactive amines (Histamine, Serotonin)
- Preformed, stored in mast cell granules
- Released immediately upon injury or allergen exposure
- Cause rapid vasodilation and increased permeability
Arachidonic acid metabolites (eicosanoids)
- Produced from cell membrane phospholipids via phospholipase A2
- Two branches:
- COX pathway → Prostaglandins (vasodilation, pain, fever) and Thromboxane A2
- Lipoxygenase pathway → Leukotrienes (LTB4 = chemotaxis; LTC4/D4/E4 = permeability, bronchoconstriction)
- NSAIDs block COX → reduce prostaglandins → reduce fever, pain, swelling
- Corticosteroids block phospholipase A2 → suppress the entire pathway
Cytokines: TNF and IL-1
- Produced by activated macrophages and dendritic cells
- Increase endothelial expression of adhesion molecules (E/P-selectin, ICAM-1)
- Drive the acute-phase response: fever, neutrophilia, acute-phase protein synthesis (CRP, fibrinogen, serum amyloid A) by the liver
- At very high levels (sepsis): hypotension, reduced cardiac contractility, DIC
Complement (C3a, C5a, C5b-9)
- C3a and C5a are anaphylatoxins - stimulate mast cell degranulation → permeability
- C5a is a potent chemoattractant for neutrophils
- C5b-9 (membrane attack complex) lyses microbes directly
Bradykinin
- Produced from kininogens by kallikrein
- Causes vasodilation, increased permeability, smooth muscle contraction, and pain
Morphologic Patterns of Acute Inflammation
The gross and microscopic appearance depends on the site and severity:
| Pattern | Features | Example |
|---|---|---|
| Serous | Watery, low-protein fluid; minimal cells | Skin blister, pleural effusion from viral pleuritis |
| Fibrinous | Fibrin-rich exudate; "bread-and-butter" appearance on serosal surfaces | Fibrinous pericarditis |
| Purulent (suppurative) | Pus (neutrophils + dead cells + microbes); walled-off = abscess | Bacterial pneumonia, appendicitis |
| Ulcer | Surface defect from sloughing of necrotic tissue | Peptic ulcer, skin ulcer in peripheral vascular disease |
Outcomes of Acute Inflammation
Three possible fates:
-
Complete resolution - the ideal outcome. The injurious agent is eliminated, edema fluid drains via lymphatics, dead cells/debris removed by macrophages, parenchymal cells regenerate. Occurs when injury is limited and tissue can regenerate.
-
Healing by fibrosis (scarring) - when there is extensive tissue destruction, or tissues incapable of regeneration (e.g., cardiac muscle), or abundant fibrin that cannot be cleared. Connective tissue replaces the damaged area.
-
Progression to chronic inflammation - when the acute response cannot resolve the injurious agent (e.g., mycobacteria, persistent irritants) or healing is impaired. Characterized by mononuclear cell infiltrate (lymphocytes, macrophages), angiogenesis, and fibrosis.
Memory Summary
STIMULUS (infection/injury)
↓
RECOGNITION (TLRs, complement activation)
↓
MEDIATOR RELEASE (histamine, eicosanoids, TNF, IL-1, complement)
↓
VASCULAR RESPONSE LEUKOCYTE RESPONSE
Vasodilation (redness, heat) Margination → Rolling (selectins)
↑ Permeability (edema, pus) → Firm adhesion (integrins/ICAM-1)
→ Transmigration (PECAM-1)
→ Chemotaxis (C5a, IL-8, LTB4)
→ Phagocytosis & killing (ROS, enzymes)
↓
OUTCOMES: Resolution / Fibrosis / Chronic inflammation
Source: Robbins & Kumar Basic Pathology, 11e - Robbins, Cotran & Kumar Pathologic Basis of Disease, 10e
This is a shared conversation. Sign in to Orris to start your own chat.