Myocardial infarction
Reading File
Finding Sources
Searching PubMed
"myocardial infarction"[MeSH Terms] AND management
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Loading Image
Loading Image
I now have all the information needed for a comprehensive, well-illustrated answer. Let me compile it.
Myocardial Infarction (MI)
MI - commonly called a "heart attack" - is the death of cardiac muscle due to prolonged ischemia. Roughly 800,000 individuals in the United States experience an MI each year (nearly one every 40 seconds), causing almost 400,000 deaths annually. The major underlying cause is atherosclerosis. - Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 511
1. Pathogenesis
Coronary Arterial Occlusion
The typical sequence of events underlying most MIs:
- An atheromatous plaque is eroded or disrupted by endothelial injury, intraplaque hemorrhage, or mechanical forces - exposing subendothelial collagen and necrotic plaque contents to blood.
- Platelets adhere, aggregate, and activate, releasing thromboxane A₂, ADP, and serotonin - causing further platelet aggregation and vasospasm.
- Activation of coagulation via tissue factor and other mechanisms adds to the growing thrombus.
- Within minutes, the thrombus completely occludes the coronary artery lumen.
When angiography is performed within 4 hours of MI onset, coronary thrombosis is demonstrated in almost 90% of cases. These thrombi are usually at a site that did not previously have a critical (>70%) fixed stenosis. - Robbins, p. 511
Less Common Causes (~10% of MI)
- Vasospasm (with or without atherosclerosis) - cocaine, ephedrine
- Embolism from mural thrombus, AF, infective endocarditis, prosthetic material
- Vasculitis, sickle cell disease, amyloid deposition, aortic stenosis, hypotension/shock
Myocardial Response to Ischemia
| Time | Event |
|---|---|
| Seconds | Cessation of aerobic metabolism; depletion of creatine phosphate and ATP |
| ~1 minute | Loss of contractility |
| Minutes | Ultrastructural changes (myofibrillar relaxation, glycogen depletion, mitochondrial swelling) - reversible |
| 20-30 min | Irreversible necrosis begins if blood flow ≤10% of normal |
| 6-12 hours | Complete loss of viability in the area at risk |
The subendocardial zone is injured first - it is the last to receive blood from epicardial vessels and is exposed to high intramural compressive pressures. The wavefront of necrosis then progresses centripetally (from subendocardium outward). - Robbins, p. 512
Cardiac muscle requires about 1.3 mL O₂/100 g/min just to remain alive; the normal resting left ventricle receives ~8 mL O₂/100 g/min. If ≥15-30% of normal resting blood flow is maintained, the muscle survives. - Guyton & Hall Textbook of Medical Physiology, p. 271
2. Patterns of Infarction
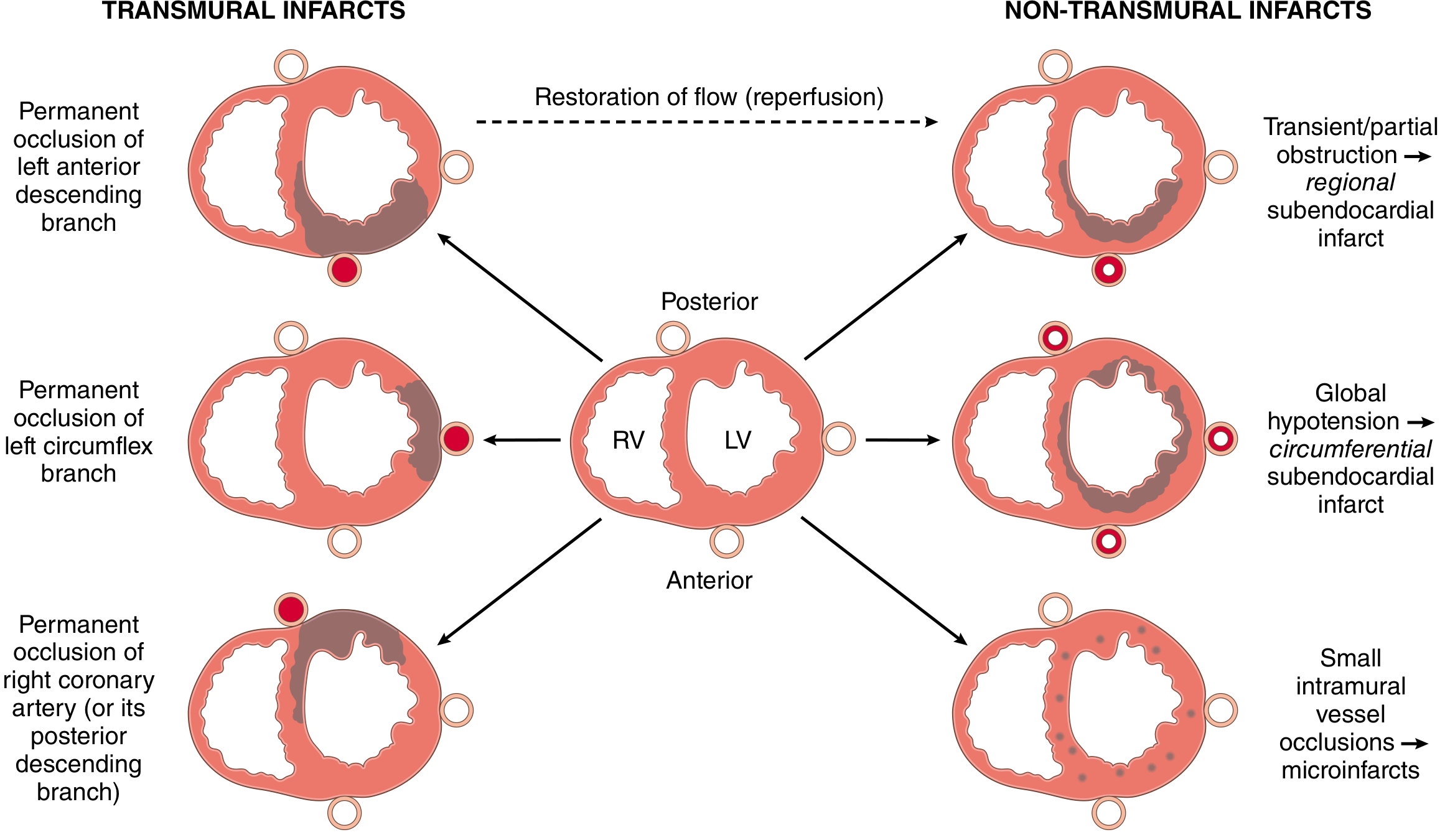
Fig. 12.11 - Robbins: Transmural infarcts (left) by coronary artery territory vs. non-transmural patterns (right)
| Pattern | Cause | Territory |
|---|---|---|
| Transmural | Full epicardial vessel occlusion (plaque + thrombus) | Full-thickness wall necrosis in one vessel territory |
| Subendocardial (regional) | Transient/partial obstruction or thrombolysis before full-thickness necrosis | Subendocardial zone in one territory |
| Subendocardial (circumferential) | Global hypotension superimposed on diffuse CAD | Subendocardial zone around entire circumference |
| Microinfarcts | Small intramural vessel occlusion (emboli, vasculitis, catecholamines) | Scattered microscopic foci |
Coronary Artery Territories
- LAD (left anterior descending): anterior wall of LV, apex, anterior 2/3 of ventricular septum
- LCX (left circumflex): lateral wall of LV
- RCA (right coronary): entire RV free wall, posterior LV, posterior 1/3 of septum (in ~80% right-dominant circulations)
3. Gross and Microscopic Pathology
Gross Appearance
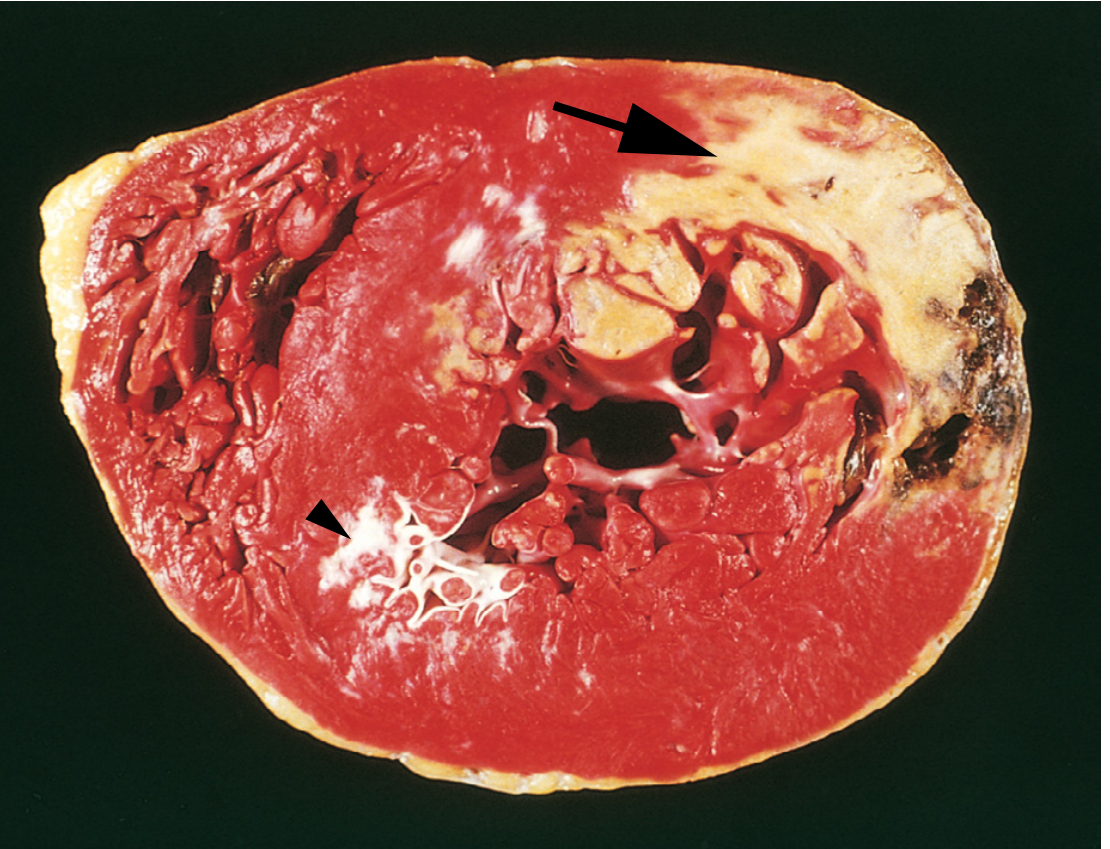
Fig. 12.12 - Robbins: TTC (triphenyl tetrazolium chloride) staining identifies viable myocardium (red) vs. necrotic areas (pale, unstained - arrow)
Grossly, an MI is undetectable in the first 6-12 hours. By 24-72 hours, a pale, poorly demarcated area appears. Later the infarct becomes yellow-tan and soft, then progressively replaced by gray-white fibrous scar over 6+ weeks.
Microscopic Progression
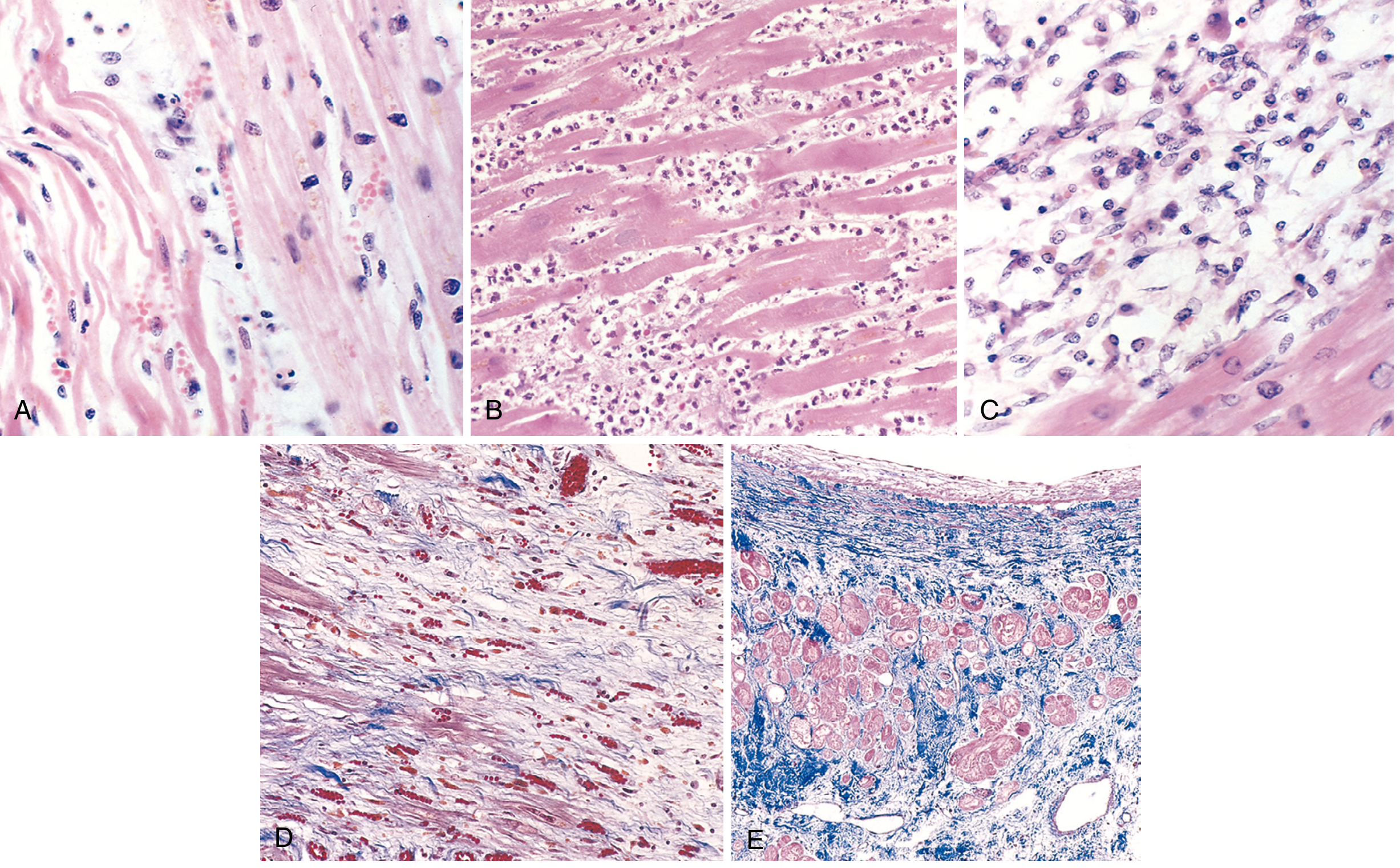
Fig. 12.13 - Robbins: Histological stages of MI
| Time | Microscopic Feature |
|---|---|
| Day 0-6 hrs | Normal - no light microscopic change |
| Day 1 | Coagulative necrosis + wavy fibers + edema; pyknotic nuclei |
| Days 1-3 | Acute inflammation: neutrophil infiltration (most prominent 1-3 days) |
| Days 3-7 | Dense neutrophil infiltrate; macrophage arrival begins |
| Days 7-10 | Macrophage phagocytosis of necrotic myocytes; granulation tissue begins |
| Weeks 2-6 | Granulation tissue (loose collagen, capillaries) |
| >6 weeks | Dense collagenous scar - electrically and mechanically inert |
Because cardiac myocytes are terminally differentiated, no cardiomyocyte proliferation occurs. Once healed, the scar looks the same whether 8 weeks or 10 years old. - Robbins, p. 514-515
4. Clinical Features & Classification
Symptoms
- Chest pain: crushing, pressure, squeezing; radiating to left arm, jaw, neck
- Diaphoresis (cold sweat)
- Dyspnea, nausea, vomiting
- Sense of doom
- Silent MI in diabetics and the elderly (up to 20-30% of cases)
ECG Classification
| Type | ECG Finding | Pathology |
|---|---|---|
| STEMI | ST-segment elevation in leads over the infarct | Transmural ischemia - usually complete occlusion |
| NSTEMI | No ST elevation; ST depression or T-wave changes | Subendocardial or partial-thickness ischemia |
ECG Mechanism of ST Elevation (Ganong's Review of Medical Physiology, p. 534):
Three simultaneous abnormalities cause ST elevation:
- Rapid repolarization in infarcted fibers (accelerated K⁺ channel opening) → current flows out of infarct → ST elevation
- Decreased resting membrane potential (K⁺ loss) → TQ depression recorded as apparent ST elevation
- Delayed depolarization → infarcted area relatively positive during early repolarization → ST elevation
After weeks, the ST changes resolve and Q waves appear - indicating electrically silent dead tissue (transmural infarct).
5. Biomarkers
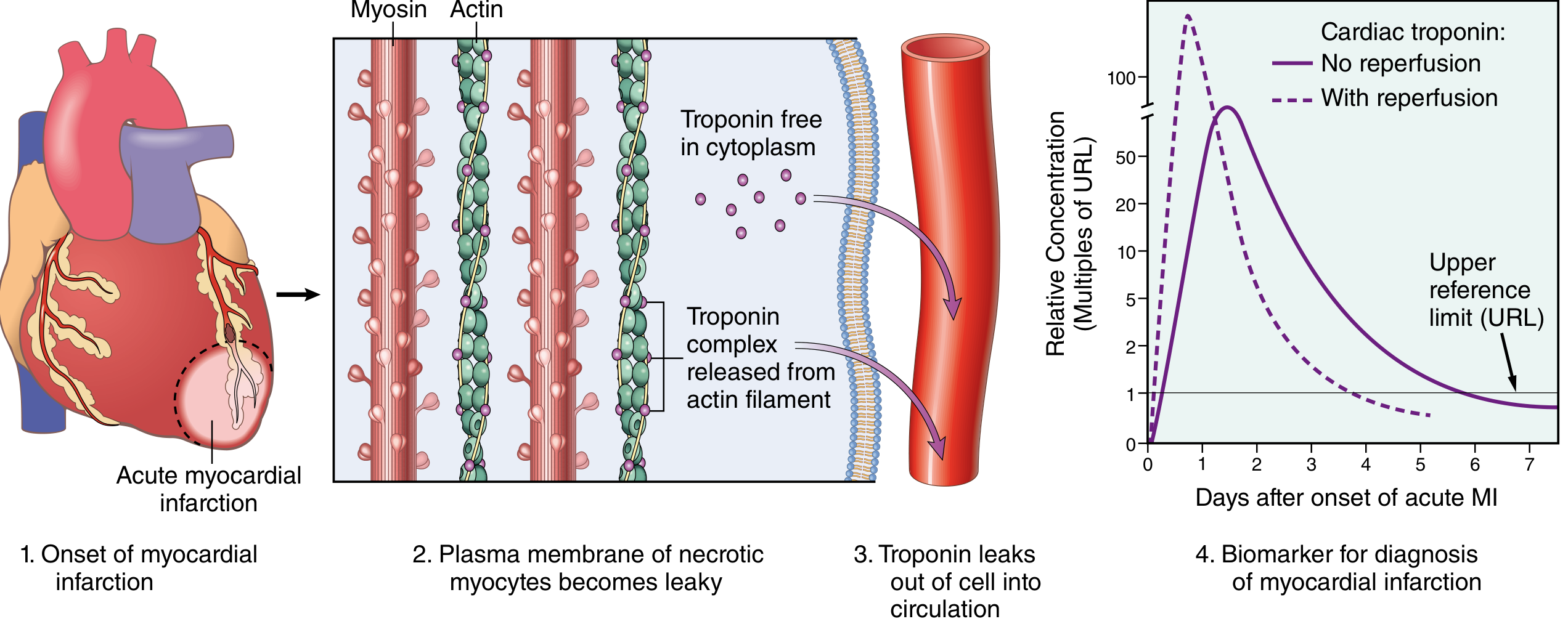
Fig. 12.16 - Robbins: Troponin I/T release kinetics. With reperfusion: earlier, higher peak (rapid washout). Without reperfusion: delayed, lower, sustained elevation.
| Biomarker | Rise | Peak | Duration |
|---|---|---|---|
| Troponin I/T (preferred) | 2-4 hrs | 24-48 hrs | 7-10 days |
| CK-MB | 3-6 hrs | 12-24 hrs | 2-3 days |
Important: "Troponin leak" (low-level elevation) occurs in CHF, pulmonary embolus, renal failure, sepsis, and myocarditis - these do not follow the typical rise-and-fall kinetics of MI. Serial measurements are essential. - Robbins, p. 517
6. Causes of Death
The most common causes of early death after acute MI (Guyton & Hall, p. 271):
- Decreased cardiac output - systolic stretch: the infarcted zone bulges outward during systole instead of contracting, worsening overall pump function ("cardiogenic shock")
- Pulmonary edema - damming of blood in pulmonary vessels; LV failure
- Ventricular fibrillation - the most common cause of sudden death in the first hour
- Cardiac rupture - free wall, interventricular septum, or papillary muscle rupture (typically days 3-7)
7. Complications
Nearly three-quarters of patients experience one or more complications after acute MI. Overall in-hospital mortality is <7% (STEMI: ~9%, NSTEMI: ~6%). One-third of out-of-hospital STEMI patients die - usually within 1 hour from arrhythmia. - Robbins, p. 517
| Complication | Timing | Notes |
|---|---|---|
| Arrhythmias | Hours-days | Most common; VF commonest cause of early death |
| Cardiogenic shock | Hours | >40% LV necrosis; high mortality |
| Acute heart failure / pulmonary edema | Hours-days | LV dysfunction |
| Free wall rupture | Days 3-7 | Cardiac tamponade; often fatal |
| Papillary muscle rupture | Days 2-7 | Acute mitral regurgitation |
| Ventricular septal rupture | Days 3-7 | Acute VSD; step-up O₂ in RV |
| Mural thrombus | Days-weeks | Embolic risk; anticoagulation needed |
| Ventricular aneurysm | Weeks-months | Paradoxical pulsation; arrhythmia risk |
| Pericarditis (Dressler syndrome) | 2-10 weeks | Autoimmune; fever, pericardial rub |
| Chronic heart failure | Months-years | Infarct expansion and remodeling |
8. Treatment
Initial management targets the underlying pathophysiology (Robbins, p. 517):
| Intervention | Goal |
|---|---|
| Oxygen | Correct hypoxia |
| Nitrates | Vasodilation, reverse vasospasm |
| Antiplatelet therapy | Aspirin + ADP-receptor inhibitors (clopidogrel/ticagrelor/prasugrel) + GPIIb/IIIa inhibitors |
| Anticoagulation | UFH, LMWH, direct thrombin inhibitors, or factor Xa inhibitors |
| Beta-blockers | Reduce O₂ demand, prevent arrhythmias (avoid in acute HF) |
| Reperfusion | Primary PCI (preferred if <120 min); thrombolytics if PCI unavailable (<30 min) |
| ACE inhibitors/ARBs | Post-MI remodeling prevention |
| Statins | Plaque stabilization, secondary prevention |
The benefits of reperfusion are greatest when achieved quickly. This is the rationale for the "time is muscle" principle: every minute of occlusion causes progressive, centripetally expanding necrosis. - Robbins, p. 512
Recent Evidence Note
A 2025 meta-analysis (PMID 39298680) in European Journal of Preventive Cardiology examined beta-blockers for secondary prevention in MI patients without reduced ejection fraction or heart failure, finding that routine long-term beta-blockade in this group may not carry the same mortality benefit previously assumed - a clinically important nuance to the traditional guideline-based recommendation.
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease (p. 511-517); Ganong's Review of Medical Physiology, 26th ed. (p. 534); Guyton & Hall Textbook of Medical Physiology (p. 271)
National immunization schedule with easy mnemonics
Reading File
Searching the Web
India National Immunization Schedule 2024 2025 UIP updated vaccines
Finding Sources
Reading Web Page
https://saplinghospitals.com/blogs/iap-immunization-schedule…
Reading File
Reading File
I now have all the data I need. Let me compile the complete, mnemonic-rich answer.
National Immunization Schedule (India) - with Mnemonics
Two schedules exist side by side in India: the Government UIP/NIS (free, at all government centres) and the IAP schedule (broader, for private practice). Both are exam-relevant.
Part 1 - Government NIS / Universal Immunization Programme (UIP)
The Master Mnemonic: "BIRTH → 6 → 10 → 14 → 9M → 16-24M → 5-6Y → 10Y → 16Y"
Think of it as "B-6-10-14-9-16-5-10-16" → "Baby, 6-10-14 weeks, 9 months, 16 months, 5 yrs, 10 yrs, 16 yrs"
AT BIRTH - Mnemonic: "Born Happy, Brave"
| Vaccine | Full Name | Route/Site |
|---|---|---|
| BCG | Bacillus Calmette-Guérin | ID, left arm |
| Hep B-0 | Hepatitis B birth dose | IM, anterolateral thigh |
| bOPV-0 | Bivalent Oral Polio Vaccine | Oral |
"B-H-b" = "Baby Has Been born" (BCG, HepB, bOPV)
6 WEEKS - Mnemonic: "PPFR + PCV = 5 Pricks at 6"
| Vaccine | Mnemonic |
|---|---|
| Pentavalent-1 (DPT + HepB + Hib) | |
| bOPV-1 | |
| fIPV-1 (fractional IPV) | |
| RVV-1 (Rotavirus) | |
| PCV-1 (Pneumococcal) |
Easy: "5 P's at 6 weeks" = Penta, bOPV, IPV, Rota, PCV → "POP-IR-P" → "Papa's Oral Polio-IPV-Rota-PCV"
10 WEEKS - Mnemonic: "PPR - No Fractions, No PCV"
| Vaccine |
|---|
| bOPV-2 |
| Pentavalent-2 |
| RVV-2 |
"10 weeks = 3 vaccines (OPV + Penta + Rota)" - Drop fIPV and PCV at 10 weeks (they return at 14 weeks)
Memory trick: "10 weeks is lean" = POP-R only (Penta, OPV, Rota)
14 WEEKS - Mnemonic: "Same as 6, minus one R"
| Vaccine |
|---|
| bOPV-3 |
| Pentavalent-3 |
| fIPV-2 |
| RVV-3 (last dose of Rota) |
| PCV-2 |
14 weeks = same 5 vaccines as 6 weeks (IPV and PCV return, Rota completes its series)
Memory trick: "6 weeks = 14 weeks. 10 weeks is the odd one out"
9-11 MONTHS - Mnemonic: "MJP + Vit A" = "Major Junction Point"
| Vaccine | Note |
|---|---|
| MR-1 (Measles-Rubella) | |
| JE-1* (Japanese Encephalitis) | *Endemic districts only |
| fIPV-3 | 3rd fractional IPV dose |
| PCV Booster | |
| Vitamin A - 1st dose (1,00,000 IU) |
Mnemonic: "9 months = MJP Vitamin" → "My Junior Passed with Vitamins" (MR, JE, IPV, PCV, Vit A)
16-24 MONTHS - Mnemonic: "MJ-DPT BOOster + Vit A"
| Vaccine |
|---|
| MR-2 |
| JE-2* |
| DPT Booster-1 |
| bOPV Booster |
| Vitamin A (2nd dose, 2,00,000 IU) - then every 6 months till 5 years |
Mnemonic: "16 months = MJD-BO-VA" → "My Junior Doctor BOosts Vitamins Again"
5-6 YEARS
DPT Booster-2 only
Mnemonic: "5 years = DPT alone" → "DPT Departs at 5"
10 YEARS & 16 YEARS
Td (Tetanus + low-dose diphtheria) at both ages
Mnemonic: "Ten & Sixteen = Td Twice as Teen"
PREGNANT WOMEN - Mnemonic: "TT-1 TT-2 or Booster"
| Visit | Vaccine |
|---|---|
| First ANC visit (or at registration) | Td-1 |
| 4 weeks after Td-1 | Td-2 |
| If previously immunised (last 3 years) | Td Booster (single dose) |
Mnemonic: "Pregnant = Two Td's, or One Booster"
Complete NIS Quick Reference Table
| Age | Vaccines | Mnemonic |
|---|---|---|
| Birth | BCG, HepB-0, bOPV-0 | Born Happy Baby |
| 6 weeks | Penta-1, bOPV-1, fIPV-1, RVV-1, PCV-1 | Papa's Oral Polio Is Really Preventive |
| 10 weeks | Penta-2, bOPV-2, RVV-2 | POR (lean week) |
| 14 weeks | Penta-3, bOPV-3, fIPV-2, RVV-3, PCV-2 | Same as 6 weeks (mirror image) |
| 9-11 months | MR-1, JE-1*, fIPV-3, PCV booster, Vit A | MJ iP Vitamin |
| 16-24 months | MR-2, JE-2*, DPT-B1, bOPV-B, Vit A | MJD-BO-VA |
| 5-6 years | DPT Booster-2 | DPT Departs at 5 |
| 10 years | Td | Ten = Td |
| 16 years | Td | Sweet Sixteen = Td |
| Pregnancy | Td-1, Td-2 / Td Booster | Two Td's or Booster |
Part 2 - IAP Schedule (Private Practice / Broader Coverage)
The IAP schedule adds vaccines not in the government UIP due to cost constraints. - Park's Textbook of Preventive & Social Medicine
Mnemonic for EXTRA IAP vaccines: "MATH-VHI"
| Vaccine | Age |
|---|---|
| MMR | 15 months (instead of just MR) |
| AHepatitis A | 12 months + 18 months (2 doses) |
| Typhoid Conjugate | 9 months, then booster |
| HPV | 9-14 years (girls) |
| Varicella | 15 months + 4-6 years |
| Hib (separate) | If not using Pentavalent |
| Influenza | 6 months, annually |
IAP Schedule Summary
| Age | IAP Vaccines |
|---|---|
| Birth | BCG, OPV-0, HepB-1 |
| 6 weeks | DPT-1, IPV-1, Hib-1, HepB-2, Rota-1, PCV-1 |
| 10 weeks | DPT-2, IPV-2, Hib-2, Rota-2, PCV-2 |
| 14 weeks | DPT-3, IPV-3, Hib-3, Rota-3, PCV-3 |
| 6 months | HepB-3 |
| 9 months | MMR-1, Typhoid Conjugate |
| 12 months | Hepatitis A-1 |
| 15 months | MMR-2, Varicella-1, PCV Booster |
| 18 months | DPT Booster-1, Hib Booster |
| 2 years | Hepatitis A-2 |
| 4-6 years | DPT B-2, IPV-B, MMR-3, Varicella-2 |
| 10-12 years | Tdap, HPV (girls) |
Part 3 - Key Exam Points with Mnemonics
Pentavalent Vaccine = "DPT + Hep B + Hib" = "5-in-1"
- Replaces DPT, HepB, and Hib separately in the government schedule
- Given at 6, 10, 14 weeks
fIPV (Fractional IPV) = "Given 3 times" at 6 weeks, 14 weeks, 9-11 months
- Fractional = 1/5th of the full dose, given intradermal
- Mnemonic: "6, 14, 9" = "FIX-teen-9"
Vitamin A Supplementation (in NIS):
- 1st dose: 1,00,000 IU at 9 months with MR-1
- 2nd-9th doses: 2,00,000 IU every 6 months from 16 months to 5 years
- Mnemonic: "9 months = 1 lakh; after that = 2 lakhs every 6 months till 5"
Rotavirus Vaccine (RVV):
- 3 doses: 6 weeks, 10 weeks, 14 weeks → "6-10-14 only" - series ends at 14 weeks
- Mnemonic: "Rota stops at 14"
JE Vaccine:
- Only in endemic districts
- 2 doses: 9-11 months and 16-24 months
- Mnemonic: "JE = Just Endemic" (not universal)
Big Picture Memory Aid - "BOMB the Polio Rota PCV Axis"
| Letter | Week |
|---|---|
| B = Birth | BCG, bOPV, HepB |
| 6 | +fIPV +RVV +PCV +Penta |
| 10 | Drop IPV & PCV |
| 14 | IPV & PCV return |
| 9M | MR + JE + PCV booster + Vit A |
| 16-24M | MR + JE + DPT boost + OPV boost + Vit A |
Booster Dose Summary - Mnemonic: "DPT Boosts Twice, OPV Once, Td Twice as Teen"
| Vaccine | Booster Ages |
|---|---|
| DPT | 16-24 months (B1), 5-6 years (B2) |
| OPV | 16-24 months (once) |
| PCV | 9-11 months (once) |
| Td | 10 years, 16 years |
| MR | 16-24 months (= dose 2, also counts as booster) |
Sources: India NIS - Government of India / CoWIN; Park's Textbook of Preventive and Social Medicine; IAP Advisory Committee on Vaccines and Immunization Practices (ACVIP)
This is a shared conversation. Sign in to Orris to start your own chat.