Heart failure
Asking for Preferences
Reading File
Finding Sources
Searching PubMed
"heart failure" AND pathophysiology
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Here is a comprehensive overview of heart failure pathophysiology, drawn from multiple authoritative textbooks.
Heart Failure: Overview & Pathophysiology
Definition & Basic Concept
Heart failure (HF) is a clinical syndrome in which the heart cannot pump sufficient blood to meet the metabolic demands of the body (forward failure), or can only do so at abnormally elevated filling pressures (backward failure), or both. Normal cardiac output is approximately 5 L/min in a 70 kg adult.
Types of Heart Failure
| Type | Mechanism | EF |
|---|---|---|
| Systolic (HFrEF) | Reduced contractility / pump failure | < 45% (normal >60%) |
| Diastolic (HFpEF) | Stiff, non-compliant ventricle; impaired relaxation | Preserved (≥50%) |
| High-output failure | Demands exceed even elevated output (hyperthyroidism, beriberi, AV shunts, anemia) | Often normal/elevated |
Causes & Initial Triggers
The three major triggers all increase cardiac work:
- Pressure overload → hypertension, aortic stenosis → concentric hypertrophy (new sarcomeres added in parallel)
- Volume overload → valvular regurgitation → eccentric hypertrophy/dilation (sarcomeres added in series)
- Myocardial loss → ischemic heart disease (MI) → regional dysfunction, compensatory hypertrophy of surrounding myocardium
Pathological Hypertrophy → Failure
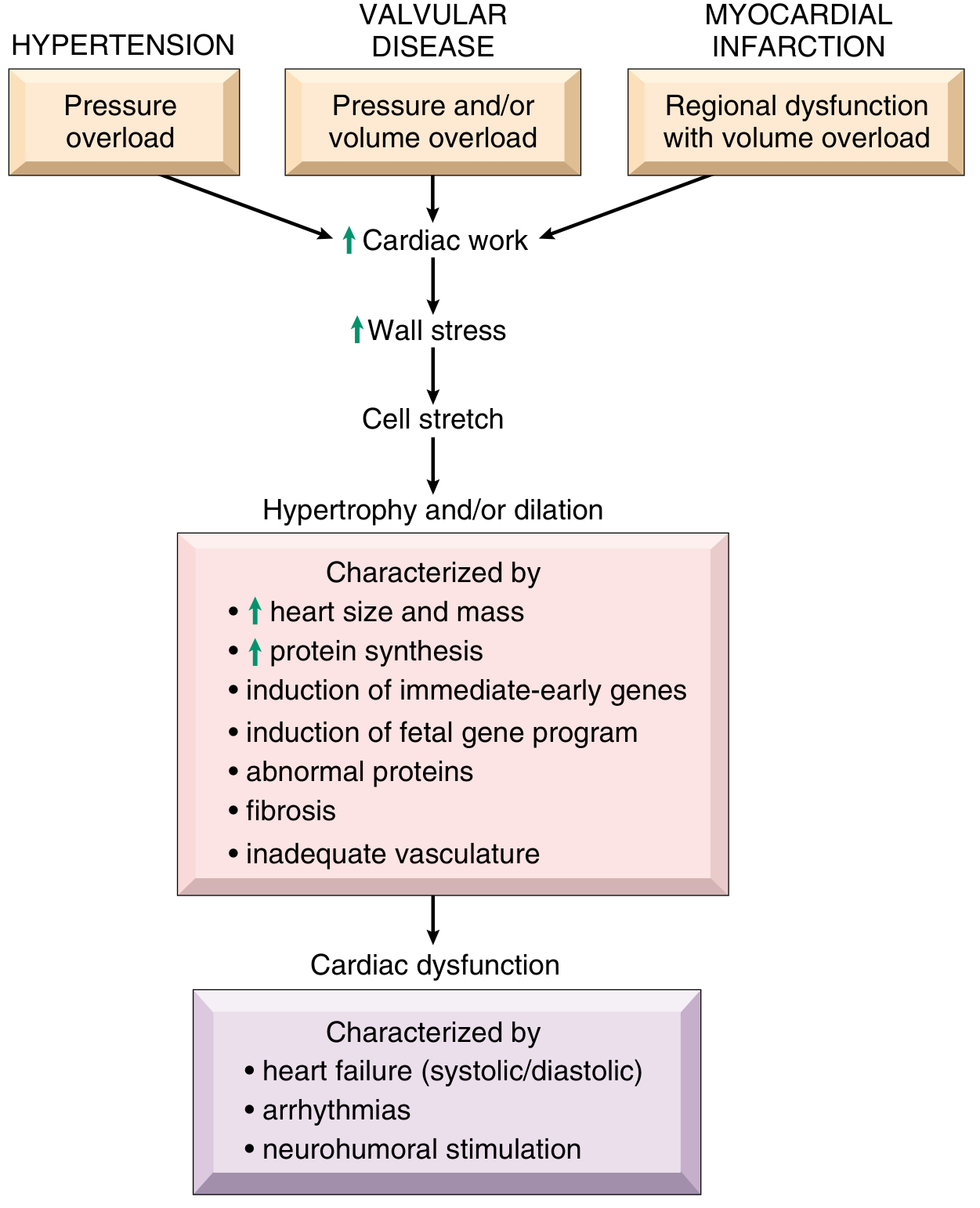
Increased cardiac work → ↑ wall stress → cell stretch → hypertrophy and/or dilation, characterized by:
- ↑ heart size and mass
- ↑ protein synthesis
- Induction of immediate-early genes (FOS, JUN, MYC, EGR1)
- Induction of a fetal gene program (fetal myosin isoforms, natriuretic peptides, collagen)
- Fibrosis and inadequate vasculature (capillary density does not increase proportionally)
Over time, this initially adaptive response becomes maladaptive, culminating in cardiac dysfunction: systolic/diastolic failure, arrhythmias, and neurohumoral stimulation.
Myocyte hypertrophy is not accompanied by proportional capillary proliferation — leading to relative ischemia in hypertrophied myocardium. — Robbins, Cotran & Kumar Pathologic Basis of Disease
Neurohumoral Compensation (The Vicious Cycle)
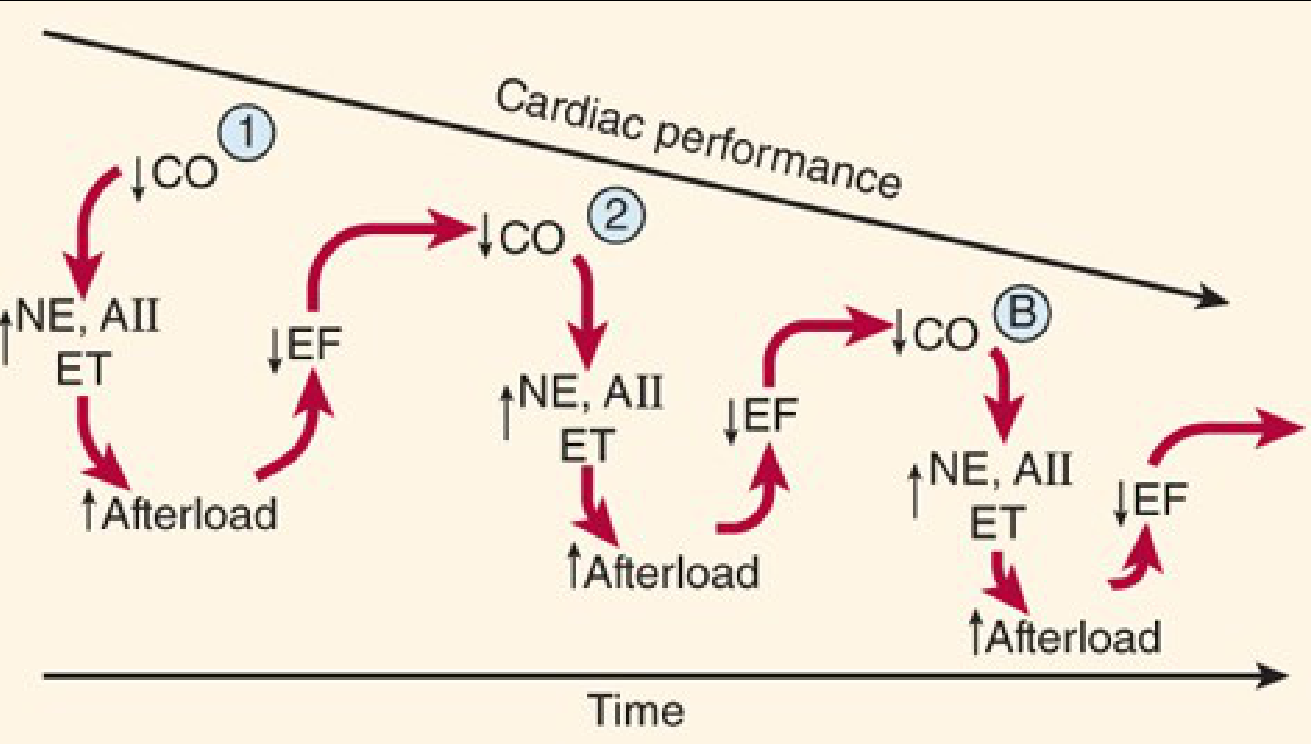
Reduced cardiac output triggers two major compensatory systems:
1. Sympathetic Nervous System
- Baroreceptors reset at lower sensitivity → ↑ sympathetic outflow, ↓ parasympathetic outflow
- → Tachycardia, ↑ contractility, vasoconstriction (↑ afterload)
- Chronic β₁ activation → receptor downregulation, Ca²⁺ leak from SR via RyR channels, ↑ apoptosis via caspases
- β₂ receptors are not downregulated; β₃ receptors (not downregulated) may mediate negative inotropic effects
2. Renin-Angiotensin-Aldosterone System (RAAS)
- ↓ Renal perfusion → ↑ renin → ↑ angiotensin II → ↑ aldosterone → sodium and water retention → ↑ preload and afterload
- Angiotensin II also promotes cardiac and vascular remodeling (fibrosis)
- Endothelin (from vascular endothelium) adds further vasoconstriction
The spiral: ↓ CO → ↑ NE/AII/Endothelin → ↑ afterload → ↓ EF → ↓ CO (repeat, progressively worsening over time)
Cellular & Molecular Changes in Heart Failure
| Change | Consequence |
|---|---|
| ↑ Immediate-early genes (FOS, JUN, MYC) | Abnormal protein synthesis |
| Fetal gene re-expression | Inefficient contractile proteins |
| SERCA impairment | Impaired Ca²⁺ re-uptake into SR |
| ↑ Phospholamban | Inhibits SERCA → ↓ Ca²⁺ cycling |
| RyR phosphorylation / PP1 upregulation | Ca²⁺ leak, dephosphorylation dysregulation |
| ↑ Caspases | Accelerated apoptosis of myocytes |
| Mitochondrial dysfunction | Impaired energy production |
| K⁺ channel remodeling | Arrhythmogenesis |
Determinants of Cardiac Performance
Four factors govern ventricular output, all deranged in HF:
- Preload — elevated in HF; rising filling pressure beyond ~15 mmHg produces a plateau, and >20–25 mmHg causes pulmonary congestion (Frank-Starling curve flattened)
- Afterload — increased by vasoconstriction (NE, AII, endothelin); directly reduces EF and CO
- Contractility — depressed by myocyte loss, β₁ downregulation, Ca²⁺ handling defects
- Heart rate — compensatory tachycardia, but reduces diastolic filling time
Left-Sided vs. Right-Sided Failure
Left-Sided HF
Causes: IHD, hypertension, aortic/mitral valve disease, cardiomyopathy
Backward effects (pulmonary):
- Pulmonary venous congestion → interstitial edema → alveolar edema
- Hemosiderin-laden macrophages in alveoli ("heart failure cells") — evidence of prior pulmonary edema
- Pleural effusions (transudates)
Symptoms: Dyspnea on exertion → orthopnea → paroxysmal nocturnal dyspnea → dyspnea at rest; fine basal rales; S₃ gallop (volume overload), S₄ (stiff ventricle)
Forward effects: ↓ renal perfusion → RAAS activation → azotemia; in severe cases, hypoxic encephalopathy (irritability → stupor → coma)
Right-Sided HF
Causes: Usually consequence of left-sided HF; also cor pulmonale (pulmonary hypertension), tricuspid/pulmonary valve disease
Backward effects (systemic venous):
- Hepatomegaly (nutmeg liver / centrilobular necrosis in severe cases)
- Splenomegaly
- Peripheral pitting edema (ankles, sacrum)
- Ascites
- Jugular venous distension
The cardiovascular system is a closed circuit — left-sided failure commonly progresses to global (biventricular) failure due to pulmonary hypertension and right ventricular strain.
Biomarkers
- BNP / NT-proBNP: Released from cardiac myocytes in response to wall stress; elevated in HF and used as a surrogate marker of presence and severity
- Rise parallels RAAS and sympathetic activation
Physiologic vs. Pathologic Hypertrophy
| Pathologic | Physiologic (exercise) | |
|---|---|---|
| Trigger | Pressure/volume overload, neurohormones | Regular aerobic exercise |
| Capillary density | Inadequate (relative ischemia) | Increases proportionally |
| Sarcomere pattern | Disorganized, fetal gene re-expression | Organized |
| Outcome | Progresses to failure | Beneficial; ↓ resting HR and BP |
Sources:
- Robbins, Cotran & Kumar — Pathologic Basis of Disease, p. 499–500
- Katzung's Basic and Clinical Pharmacology, 16th ed., p. 334–335
- Goodman & Gilman's The Pharmacological Basis of Therapeutics
This is a shared conversation. Sign in to Orris to start your own chat.