AKI and ATN
Reading File
Finding Sources
Finding Sources
Searching PubMed
"acute kidney injury"[MeSH Terms] AND management
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
Finding Sources
Reading File
Reading File
Loading Image
Finding Sources
Now I have enough content for a thorough response. Let me compile the comprehensive answer.
Acute Kidney Injury (AKI) and Acute Tubular Necrosis / Acute Tubular Injury (ATN/ATI)
1. Definition and Overview
Acute Kidney Injury (AKI) is a syndrome of abrupt decline in kidney function over hours to days, resulting in the retention of nitrogenous waste products (urea, creatinine) and disruption of fluid, electrolyte, and acid-base homeostasis.
Acute Tubular Injury/Necrosis (ATI/ATN) is the single most common cause of AKI, accounting for ~50% of all AKI in hospitalized patients and over half of ICU cases. The term "ATI" is now preferred by pathologists over the historical "ATN," because frank necrosis is not always present - injury may manifest as apoptosis or sublethal damage.
- Robbins Pathologic Basis of Disease (p. 864); Frameworks for Internal Medicine (p. 454)
2. KDIGO Staging of AKI
The RIFLE and AKIN criteria were harmonized into the KDIGO (Kidney Disease: Improving Global Outcomes) 2012 criteria, now the universal standard:
| Stage | Serum Creatinine Criteria | Urine Output Criteria |
|---|---|---|
| 1 | 1.5-1.9× baseline, or ≥0.3 mg/dL rise within 48 h | <0.5 mL/kg/h for 6-12 h |
| 2 | 2.0-2.9× baseline | <0.5 mL/kg/h for ≥12 h |
| 3 | ≥3.0× baseline, or sCr ≥4.0 mg/dL, or initiation of RRT, or eGFR <35 mL/min (age <18) | <0.3 mL/kg/h for ≥24 h, or anuria for ≥12 h |
Brenner and Rector's The Kidney (p. 1156)
Limitation: Serum creatinine is a functional (not damage) biomarker - it can rise in prerenal states without tubular injury, and remain normal with significant tubular injury in patients with high renal reserve. Novel biomarkers (NGAL, KIM-1, IL-18, TIMP-2, IGFBP7) are being studied to supplement creatinine-based definitions.
3. Classification of AKI by Location
Prerenal AKI
- Reduced renal perfusion (hypovolemia, cardiogenic shock, septic shock, RAAS activation, renal artery stenosis)
- Functional/reversible - no intrinsic tubular damage yet
- FENa <1%, BUN:Cr ratio >20, urine specific gravity >1.020
Intrarenal AKI (Intrinsic)
- ATI/ATN - ischemic or nephrotoxic (most common)
- Glomerulonephritis (rapidly progressive)
- Acute interstitial nephritis (drug-induced, infection)
- Vascular (TTP/HUS, vasculitis, atheroembolic disease)
Postrenal AKI
- Urinary obstruction (BPH, stones, malignancy, papillary necrosis)
- Must be bilateral (or unilateral in solitary kidney) to cause AKI
4. Pathogenesis of ATI/ATN
Why the Proximal Tubule is Most Vulnerable
Proximal tubular cells are packed with mitochondria, depend on oxidative phosphorylation, have a large absorptive surface area, and actively concentrate toxins via transport systems. They cannot switch to anaerobic glycolysis effectively. The outer medullary segment (PST = proximal straight tubule and thick ascending limb of Henle) is particularly at risk because it operates in a relative hypoxic environment even under normal conditions.
Two Mechanisms
A. Ischemic ATI
The following cascade occurs (see diagram below):
- Loss of cell polarity: Ischemia causes redistribution of Na,K+-ATPase from the basolateral to the luminal membrane → impaired Na+ reabsorption → increased distal Na+ delivery → tubuloglomerular feedback → afferent arteriolar vasoconstriction → ↓ GFR
- Cell detachment: Injured tubular epithelial cells slough into the lumen → luminal obstruction → ↑ intratubular pressure → ↓ GFR
- Filtrate leak: Disrupted tubular epithelium allows glomerular filtrate to leak back into the interstitium → interstitial edema → ↑ interstitial pressure → further ischemia
- Inflammatory amplification: Injured cells release cytokines/chemokines → leukocyte recruitment → additional injury
- Intrarenal vasoconstriction: Via renin-angiotensin system, endothelin release ↑, nitric oxide and prostacyclin production ↓
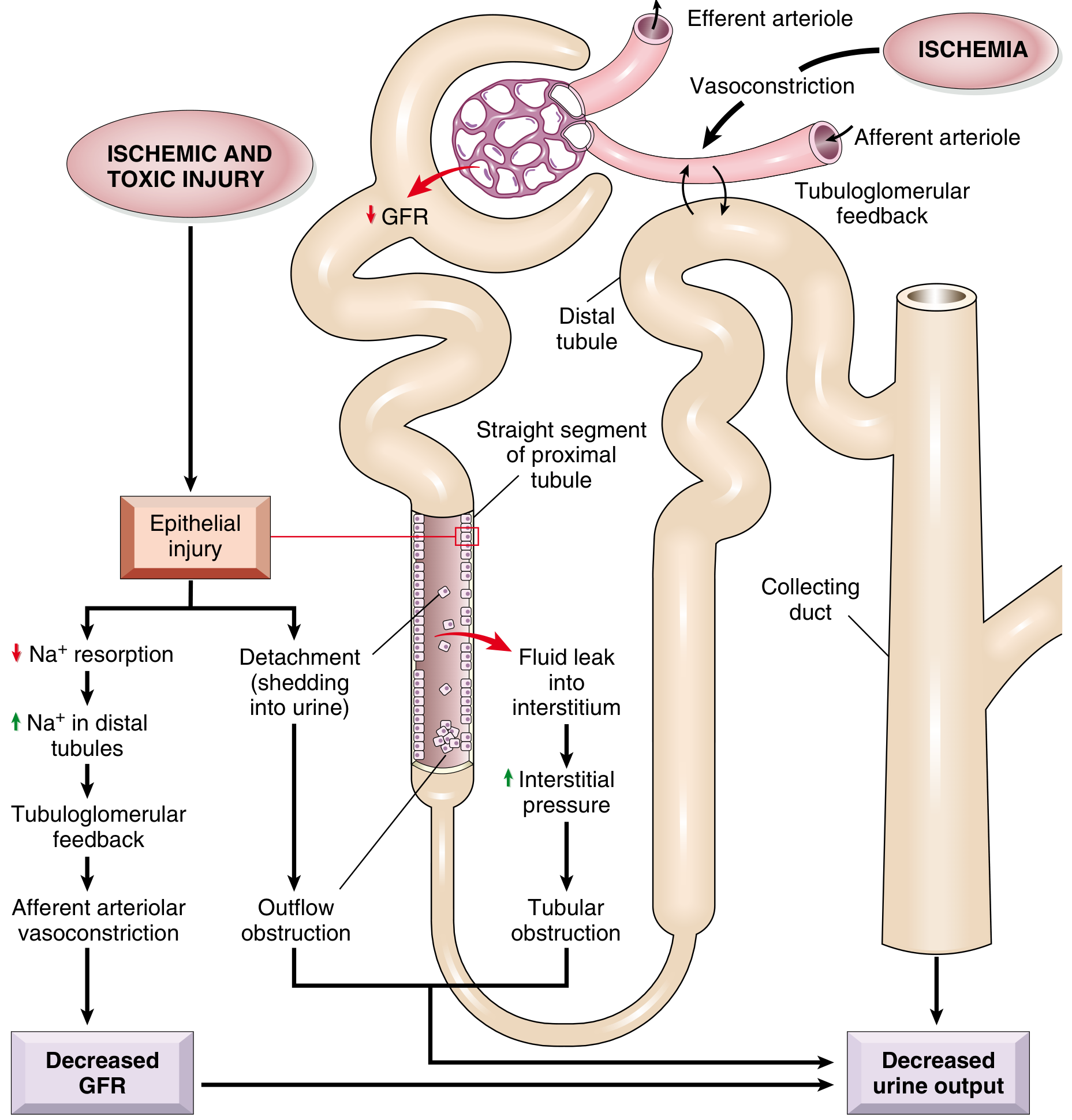
Fig. 20.28 - Postulated sequence in ischemic or toxic ATI. (Robbins Pathologic Basis of Disease)
B. Nephrotoxic ATI
- Direct toxicity to tubular epithelial cells
- Common nephrotoxins: aminoglycosides (gentamicin), IV contrast (contrast-induced nephropathy), cisplatin, amphotericin B, heavy metals (mercury), organic solvents (carbon tetrachloride), myoglobin (rhabdomyolysis), hemoglobin (hemolytic crises)
- In toxic ATI: more extensive necrosis distributed along the proximal convoluted tubule (PCT) segments (vs. patchy PST and thick ascending limb in ischemic ATI)
5. Morphology (Histopathology)
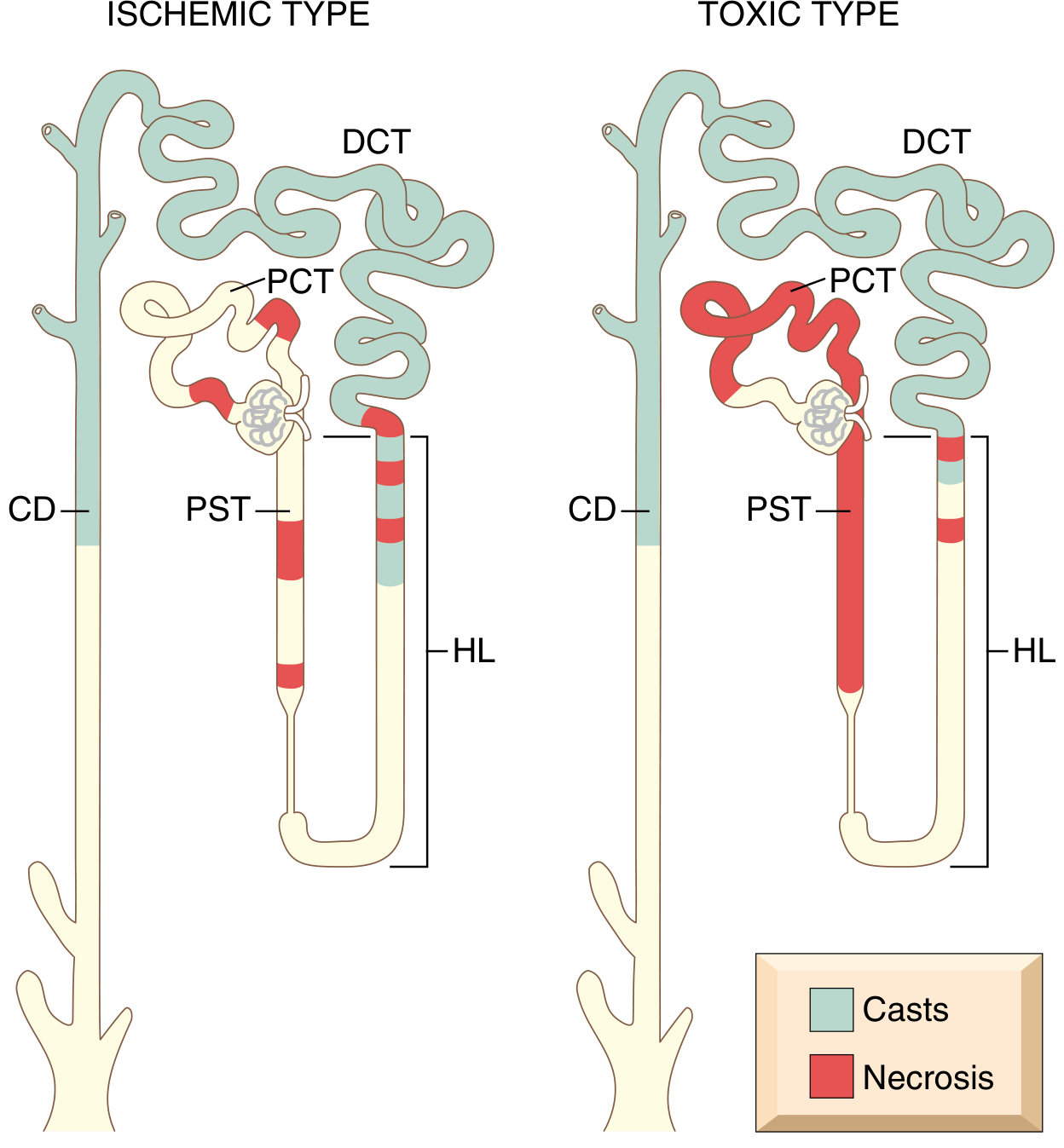
Fig. 20.29 - Patterns of tubular damage in ischemic vs toxic ATI. (Robbins, p. 864)
Ischemic ATI:
- Patchy necrosis, short segments affected
- PST (proximal straight tubule) and ascending limb of Henle loop most vulnerable
- Tubulorrhexis (basement membrane rupture)
- Interstitial edema, leukocytes in vasa recta
- Casts in DCT and collecting ducts
Toxic ATI:
- More extensive, confluent necrosis along PCT
- Basement membrane intact (key - allows repair/re-epithelialization)
- DCT and collecting ducts contain casts
Recovery: Patchy necrosis with intact basement membranes allows re-epithelialization mediated by growth factors. The severity of histologic findings often does NOT correlate with clinical severity.
- Robbins Pathologic Basis of Disease (p. 864-865)
6. Clinical Phases of ATN/AKI
| Phase | Duration | Features |
|---|---|---|
| Initiation | Hours-days | Precipitating insult; sCr begins to rise |
| Maintenance (Oliguric) | 1-2 weeks typically | Oliguria (<400 mL/day) or anuria; sCr peaks; risk of uremic complications |
| Recovery (Diuretic) | Days-weeks | Polyuria may occur as tubular function recovers before concentrating ability; risk of hypokalemia and hypovolemia |
Note: ~50% of ATN cases are non-oliguric (better prognosis).
7. Laboratory Findings
| Test | Prerenal AKI | Intrinsic AKI (ATN) |
|---|---|---|
| FENa | <1% | >1-2% |
| BUN:Cr ratio | >20 | ~10-15 |
| Urine osmolality | >500 mOsm/kg | ~300 mOsm/kg (isosthenuria) |
| Urine Na+ | <20 mEq/L | >40 mEq/L |
| Urine sediment | Hyaline casts (bland) | "Muddy brown" granular casts |
| FEUrea | <35% | >35-50% (useful when on diuretics) |
The "Muddy Brown" Cast - Hallmark of ATN

Fig. 33-6 - "Muddy brown" granular cast in ATN. ATN is highly likely when ≥6 granular casts are seen. (Frameworks for Internal Medicine, p. 455)
These casts are composed of sloughed, pigment-laden tubular epithelial cells. They form in the distal tubule and collecting duct.
- Frameworks for Internal Medicine (p. 454-455)
8. Causes of ATN - Clinical Correlation
| Clinical Scenario | Cause of ATN |
|---|---|
| Cardiogenic shock, hypotension | Prerenal spectrum → ischemic ATN |
| Amphotericin B (immunocompromised patient) | Direct nephrotoxicity |
| CT contrast + pre-existing CKD/DM | Contrast-induced nephropathy |
| Crush injury, rhabdomyolysis | Pigment nephropathy (myoglobin) |
| Hemolytic transfusion reaction | Hemoglobinuria |
| Myeloma (anemia, ↑protein gap, ↓anion gap) | Light chain (myeloma kidney) |
9. Complications of AKI (AEIOU mnemonic)
- A - Acidosis (metabolic, high anion gap)
- E - Electrolytes (hyperkalemia, hyperphosphatemia, hyponatremia, hypocalcemia)
- I - Ingestions/toxins (inability to clear)
- O - Overload (fluid - pulmonary edema)
- U - Uremia (pericarditis, encephalopathy, platelet dysfunction, nausea/vomiting)
10. Management
General Principles
- Identify and remove the cause - stop nephrotoxins, correct hypovolemia, treat sepsis, relieve obstruction
- Volume optimization - restore euvolemia with isotonic crystalloids (balanced solutions preferred over normal saline in most settings); avoid fluid overload
- Hemodynamic support - target MAP >65 mmHg; vasopressors in septic shock
- Avoid nephrotoxins - NSAIDs, ACEi/ARBs, aminoglycosides (or dose-adjust), IV contrast (use iso-osmolar, minimize dose, pre-hydrate)
- Monitor electrolytes and acid-base - treat hyperkalemia aggressively
- Nutrition - adequate protein (avoid restriction in non-dialyzed patients)
- Medication dose adjustment - renally cleared drugs
Renal Replacement Therapy (RRT) - Indications
AEIOU mnemonic above (refractory to medical management):
- Refractory hyperkalemia
- Severe/refractory metabolic acidosis (pH <7.1)
- Pulmonary edema unresponsive to diuretics
- Uremic complications (pericarditis, encephalopathy, bleeding)
- Stage 3 AKI in appropriate clinical context
No proven pharmacologic therapy exists for established ATN
- Dopamine, fenoldopam, NAC for contrast prophylaxis - not proven to reduce established ATN
- Diuretics do NOT improve outcomes in ATN (may convert oliguric to non-oliguric but don't shorten course)
- Brenner and Rector's The Kidney (pharmacologic therapy for ATN)
11. Prognosis and Recovery
ATI is generally reversible if the underlying cause is corrected and basement membranes remain intact. Recovery depends on:
- Severity and duration of the insult
- Patient age and baseline kidney function (CKD, diabetes, hypertension lower the threshold for injury)
- Whether sepsis is the precipitant (worst prognosis)
- Complications during the oliguric phase
Survivors of severe AKI have a significantly increased long-term risk of CKD, ESRD, and cardiovascular events. AKI-to-CKD transition is now a recognized entity.
Key Sources:
- Robbins, Cotran & Kumar Pathologic Basis of Disease - ATI pathogenesis and morphology (pp. 863-865)
- National Kidney Foundation Primer on Kidney Diseases, 8e - ATI mechanisms (pp. 972+)
- Brenner and Rector's The Kidney, 2-Volume Set - KDIGO staging, biomarkers, pharmacologic therapy
- Frameworks for Internal Medicine - Clinical features, urine sediment (pp. 454-455)
This is a shared conversation. Sign in to Orris to start your own chat.