Von willi brand disease
Reading File
Finding Sources
Searching PubMed
"von Willebrand disease"[MeSH Terms] AND management
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Here is a comprehensive overview of von Willebrand Disease (VWD):
Von Willebrand Disease (VWD)
Definition & Epidemiology
VWD is the most common inherited bleeding disorder, caused by quantitative or qualitative defects in von Willebrand factor (vWF). Clinically significant bleeding affects approximately 1 in 1,000 individuals, with relatively consistent prevalence across races and ethnicities. Because it is autosomal, both sexes are affected, but women are more commonly diagnosed due to greater hemostatic challenges (e.g., menstruation, childbirth).
Pathophysiology
vWF is a large multimeric plasma glycoprotein with two critical roles in hemostasis:
- Platelet adhesion — mediates platelet binding to exposed subendothelial collagen at sites of vascular injury (via platelet receptor GpIb)
- Factor VIII carrier — protects factor VIII from premature proteolytic degradation in the circulation
vWF is synthesized in endothelial cells (stored in Weibel-Palade bodies) and megakaryocytes (stored in platelet α-granules). Its gene spans 178 kb on chromosome 12 (52 exons).
The multimeric structure is key: high-molecular-weight (HMW) multimers have greater platelet-binding affinity and are most hemostically active. Multimer size is regulated by the metalloprotease ADAMTS13.
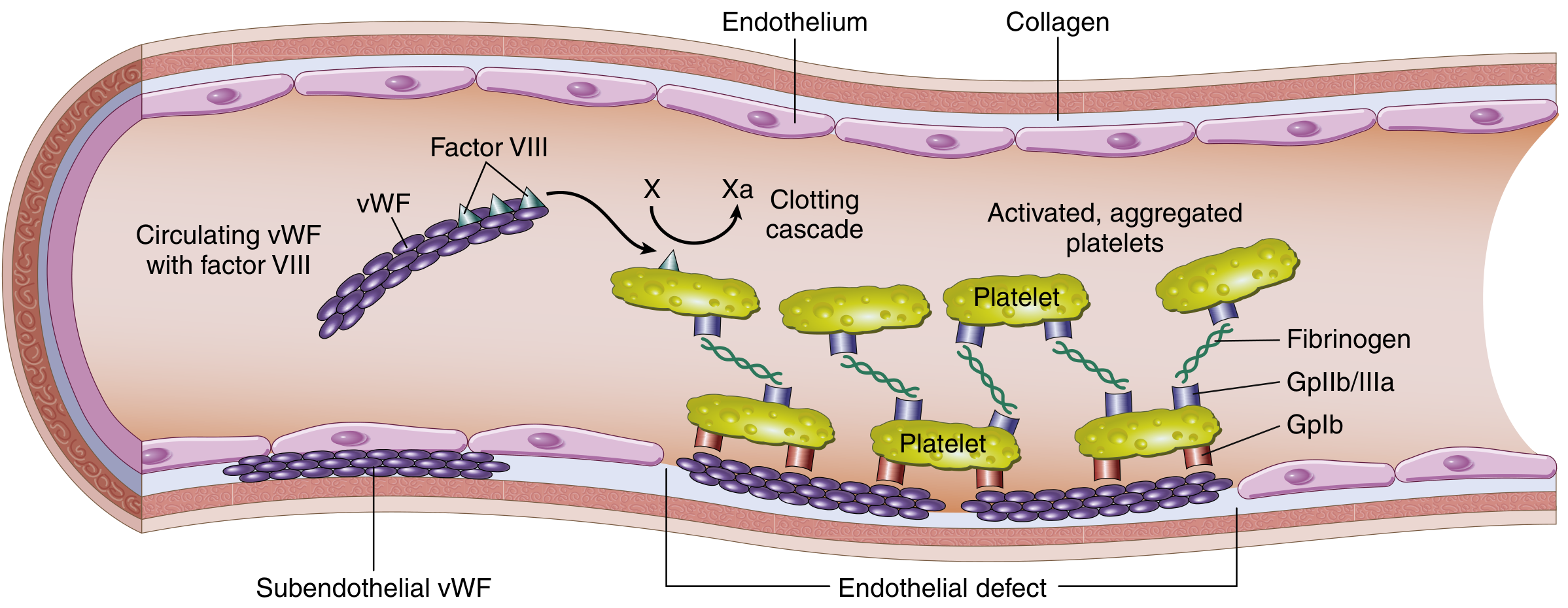
Structure and function of the Factor VIII–vWF complex: vWF circulates with Factor VIII, protects it from degradation, and mediates platelet adhesion to subendothelial collagen via GpIb. Platelet aggregation then occurs via GpIIb/IIIa and fibrinogen. — Robbins & Kumar Basic Pathology
Classification
| Type | Description | vWF:Ag | vWF Activity | Multimers |
|---|---|---|---|---|
| Type 1 | Partial quantitative deficiency (most common, ~75%) | ↓ (<30%) | ↓ proportionally | Normal distribution, reduced quantity |
| Type 2A | Qualitative — loss of HMW multimers | Normal to ↓ | ↓↓ | HMW forms absent (synthesis defect) |
| Type 2B | Qualitative — gain-of-function; HMW multimers cleared rapidly | Normal to ↓ | ↓↓ | HMW forms absent; spontaneous platelet aggregation; may cause thrombocytopenia |
| Type 2M | Qualitative — reduced platelet adhesion, HMW multimers intact | Normal to ↓ | ↓ | Normal |
| Type 2N | Qualitative — reduced Factor VIII binding only | Normal | Normal | Normal (FVIII markedly ↓) |
| Type 3 | Near-complete absence of vWF (autosomal recessive, severe) | <3% | <3% | Absent |
| "Low vWF" | Borderline levels (30–50 IU/dL), may not have true gene mutation | 30–50% | 30–50% | Normal |
Note on ABO blood group: Blood group O individuals have ~25% lower vWF levels than non-O individuals, which can affect diagnosis at borderline levels.
Clinical Manifestations
Primarily mucocutaneous bleeding:
- Epistaxis (nosebleeds)
- Easy bruising / ecchymosis
- Gum bleeding
- Prolonged bleeding from minor wounds
- Heavy menstrual bleeding (up to 80% of women with VWD)
- Recurrent GI bleeding (more common in types 2 and 3)
Type 3 and some type 2 patients may also have hemarthroses and deep tissue bleeding (due to severely low Factor VIII, resembling hemophilia).
Diagnosis
Laboratory tests:
| Test | Finding in VWD |
|---|---|
| PT | Usually normal |
| aPTT | May be prolonged (if FVIII ↓) |
| vWF antigen (vWF:Ag) | ↓ in types 1, 3; normal/↓ in type 2 |
| vWF ristocetin cofactor activity (RCo) | ↓ in most types |
| vWF:RCo / vWF:Ag ratio | <0.7 suggests qualitative defect (type 2) |
| vWF multimer analysis | Distinguishes 2A/2B/2M |
| Ristocetin-induced platelet aggregation (RIPA) | ↑ at low doses in type 2B |
| vWF:FVIII binding assay | ↓ in type 2N |
| vWFpp:vWF:Ag ratio | ↑ in type 1C (increased clearance) |
Diagnosis requires bleeding phenotype + abnormal labs, ideally repeated on two occasions weeks apart. A standardized Bleeding Assessment Tool (BAT) is used: scores ≥4 in males or ≥6 in females warrant investigation.
Treatment
1. DDAVP (Desmopressin) — First-line for responsive patients
- Synthetic vasopressin analog that triggers vWF release from Weibel-Palade bodies
- IV: 0.3 µg/kg in 100 mL saline over 20 min (max 25–30 µg)
- Intranasal: 300 µg (or 150 µg/nostril for >50 kg)
- Effective: Types 1, 2A, 2M (some patients)
- Avoid in Type 2B (triggers thrombocytopenia) and Type 3 (no stores to release)
- Type 1C patients also do NOT respond well (increased clearance)
- Tachyphylaxis develops with repeated doses; can repeat every 12–24 hr acutely
2. VWF Replacement (Plasma-derived or Recombinant VWF Concentrate)
- Used when DDAVP is contraindicated/ineffective, or for major bleeding/surgery
- Initial dose: 40–60 RCo units/kg; Maintenance: 20–40 RCo units/kg every 8–24 hr
- Target troughs: >50% RCo; peaks <200% RCo, <250% FVIII
- Duration: 7–14 days for major surgery; 1–5 days for minor surgery
3. Tranexamic Acid (antifibrinolytic)
- Oral: 15–25 mg/kg three times daily; IV: 15 mg/kg three times daily
- Useful adjunct especially for dental procedures (7–10 days) and menorrhagia
- Avoid in significant hematuria (risk of clot retention/urinary obstruction)
- Minimal thrombotic risk even in high-risk populations
4. Hormonal therapy — Oral contraceptives for heavy menstrual bleeding
Key Points for Type 2B
Type 2B has a gain-of-function mutation causing spontaneous, inappropriate platelet aggregation by the abnormal HMW multimers — similar in mechanism to TTP. This depletes both HMW multimers AND platelets, causing mild chronic thrombocytopenia. DDAVP is contraindicated (would release more "hyperfunctional" multimers, worsening thrombocytopenia).
Sources: Goldman-Cecil Medicine, 26th ed. (Ch. 159); Robbins & Kumar Basic Pathology; Henry's Clinical Diagnosis and Management by Laboratory Methods
This is a shared conversation. Sign in to Orris to start your own chat.