Identify the typical age, location, imaging, and histopathologic features of major central nervous system tumors, including meningioma, medulloblastoma, astrocytoma, ependymoma, oligodendroglioma, and glioblastoma.
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
I now have all the information needed for a comprehensive answer. Let me compile it.
Major CNS Tumors: Age, Location, Imaging, and Histopathology
Below is a systematic breakdown of each tumor, synthesized from Robbins & Cotran Pathologic Basis of Disease, Robbins & Kumar Basic Pathology, Bradley and Daroff's Neurology, and Grainger & Allison's Diagnostic Radiology.
1. Meningioma
Typical Age & Demographics
- Most common intracranial tumor (>one-third of newly diagnosed nervous system tumors). Shows a moderate 2:1 female predominance in adults; the ratio is 10:1 for spinal meningiomas. Uncommon in children. Most are slow-growing and may be incidentally discovered.
Location
- 90% are supratentorial. Common sites, in decreasing frequency: parasagittal convexity, lateral cerebral convexity, sphenoid wing, olfactory groove, sella turcica, foramen magnum. Infratentorial tumors arise on the posterior surface of the petrous bones and clivus. Rare variants occur intraventricularly. In NF2, they are multiple and often coexist with spinal ependymoma and vestibular schwannoma.
Imaging
- CT: 60% are spontaneously hyperdense; up to 20% contain calcification. Intense, uniform contrast enhancement. May show hyperostotic bone reaction (distinguishing it from schwannoma, which causes bone thinning). En plaque meningiomas spread sheet-like along dura with adjacent hyperostosis.
- MRI: Frequently isointense to cerebral cortex on both T1 and T2; may be invisible without contrast. After gadolinium: vivid, homogeneous enhancement. A hallmark sign is a "dural tail" - linear enhancement extending along the adjacent dura. Extra-axial location is shown by buckling of the gray-white junction, a CSF cleft at the tumor base, and a broad dural base.
Histopathology
- Rubbery, rounded, or bosselated dural mass; compresses but does not infiltrate brain.
- Major WHO grade 1 patterns: meningothelial (clusters of epithelioid cells with indistinct cell borders); fibroblastic (spindle cells in fascicles with collagen); transitional (mixed, with many whorls - tumor cells wrapped concentrically around one another); psammomatous (predominantly concentric calcified lamellations = psammoma bodies).
- Whorled architecture and psammoma bodies are the histologic hallmarks.
- Genetics: 50-60% have NF2 gene mutations (loss of chromosome 22q12, encoding merlin). NF2-wildtype tumors carry TRAF7, KLF4, AKT1, or SMO mutations and typically involve the skull base with lower malignant risk.
- Grade 2 (atypical, ~25%): increased mitotic index, brain invasion, or chordoid/clear cell histology.
- Grade 3 (anaplastic, 1-3%): sarcoma/carcinoma-like; TERT-promoter mutation and CDKN2A deletion are diagnostic of grade 3.
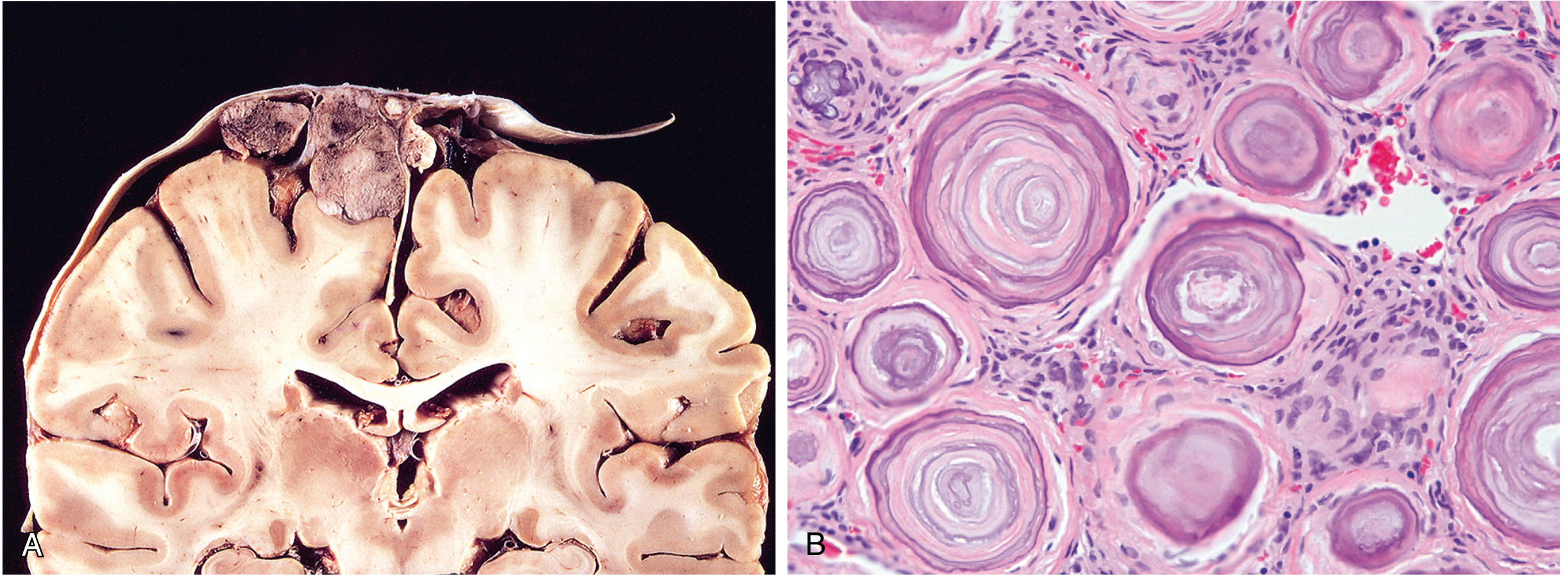
2. Medulloblastoma
Typical Age & Demographics
- Most common embryonal CNS tumor. Accounts for 20% of pediatric brain tumors. Predominantly occurs in children, though it can occur in adults (where tumors tend to be more lateral).
Location
- Exclusively in the cerebellum (by definition). In children: midline (cerebellar vermis), often near or filling the fourth ventricle - causing obstructive hydrocephalus. In adults: more lateral cerebellar hemispheres.
- Spreads to the subarachnoid space; CSF dissemination causes "drop metastases" to the cauda equina or "icing" growth along meningeal surfaces.
Imaging
- Typically a well-circumscribed, hyperdense (on CT), contrast-enhancing mass in the posterior fossa, centered on the cerebellar vermis in children.
- Often causes hydrocephalus due to fourth ventricle obstruction.
- MRI shows restricted diffusion (high cellularity) and vivid enhancement.
Histopathology
- One of the "small blue cell" tumors of childhood - densely cellular with sheets of small primitive cells, scant cytoplasm, and hyperchromatic nuclei (frequently elongated or crescent-shaped).
- Abundant mitoses; Ki-67 is high.
- Express neuronal markers (synaptophysin); glial markers (GFAP) only rarely.
- Classic morphology: Homer Wright rosettes - primitive cells arranged around a central core of delicate neuropil (no central lumen - distinguishes from ependymal rosettes).
- Desmoplastic/nodular variant: internodular desmoplasia (collagen/reticulin) with pale islands of neuronal differentiation - seen mostly in SHH-activated medulloblastoma.
- Large cell/anaplastic variant: large vesicular nuclei, prominent nucleoli, cell wrapping (cell-in-cell appearance), very high mitotic/apoptotic index.
- Molecular subtypes (WHO 2021): WNT-activated (best prognosis, ~100% 5-year survival), SHH-activated/TP53-wildtype (intermediate), SHH-activated/TP53-mutant (poor), and Groups 3 and 4. Group 3 has the worst prognosis overall.
- All subtypes are CNS WHO grade 4.
3. Astrocytoma (Diffuse, IDH-mutant) & Pilocytic Astrocytoma
A. Diffuse Astrocytoma (IDH-mutant, WHO grades 2-4)
Typical Age & Demographics
- IDH-mutant astrocytomas predominantly affect young to middle-aged adults (median age ~38 years). Presenting symptoms: seizures, headaches, focal neurologic deficits.
Location
- Cerebral hemispheres (predominantly frontal and temporal lobes); diffusely infiltrative. Grade 2 tumors expand brain parenchyma without discrete margins.
Imaging
- Grade 2: Non-enhancing, poorly defined mass with blurring of the corticomedullary junction. T2/FLAIR hyperintense. The "edema" visible on imaging often contains infiltrating tumor cells.
- Grade 3-4: Patchy enhancement as the blood-brain barrier becomes disrupted. Grade 4 IDH-mutant may show ring enhancement but is distinct from glioblastoma.
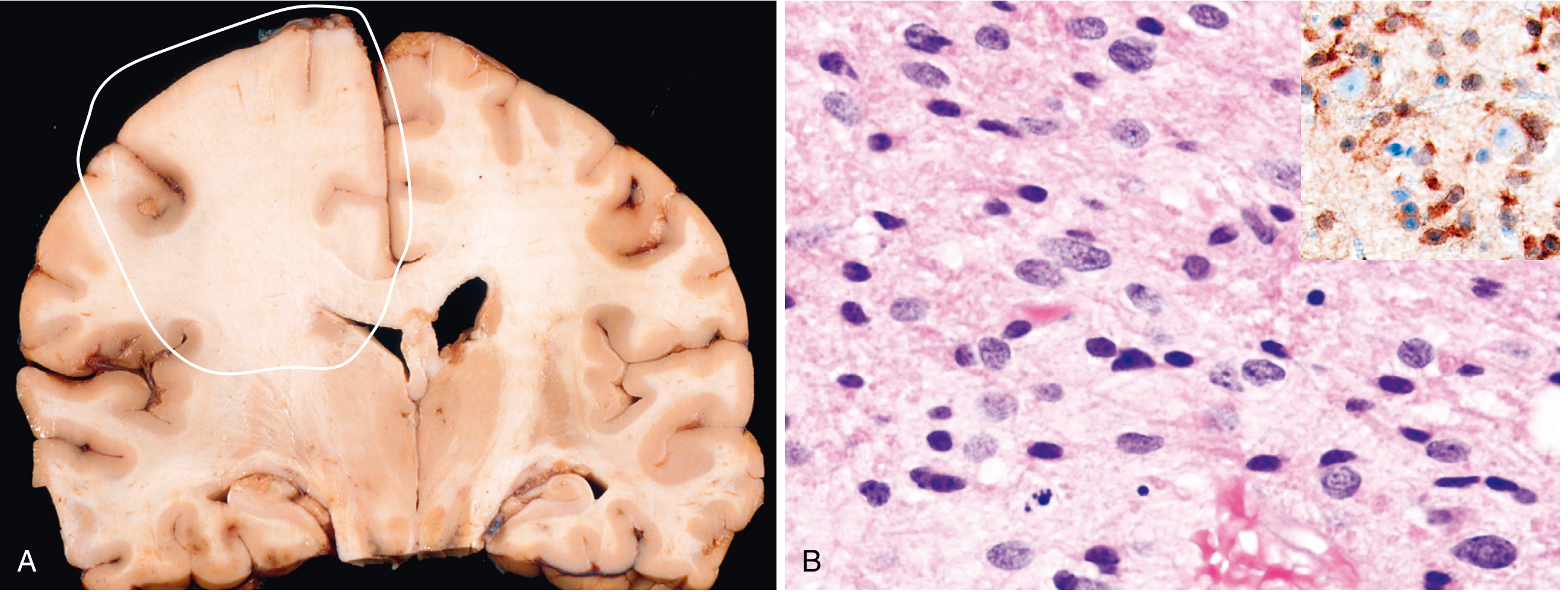
Histopathology
- Poorly defined, gray, infiltrative tumor. Firm or gelatinous; may show cystic degeneration.
- Microscopically: hypercellular compared to normal white matter; enlarged, elongated or irregular, hyperchromatic nuclei within a fibrillar GFAP-positive background.
- Hallmark: individual cells infiltrate brain tissue well beyond the main lesion ("perineuronal satellitosis", subpial spread, perivascular spread).
- IDH1 R132H immunostain positive in up to 90%. Nearly all also show ATRX loss and p53 overexpression by IHC.
- Grade 2: nuclear atypia only. Grade 3: mitoses added. Grade 4 (IDH-mutant): microvascular proliferation (MVP), necrosis, and/or homozygous CDKN2A deletion.
- Genetics: IDH1 or IDH2 mutation + TP53 mutation + ATRX inactivation (the triad).
B. Pilocytic Astrocytoma (WHO grade 1)
Typical Age & Demographics
- Children and young adults. Most common intramedullary tumor in children (up to 90% of pediatric spinal cord tumors; pilocytic subtype particularly affects ages 1-5).
Location
- Predominantly cerebellum (most common). Also: third ventricle, optic pathways (optic glioma), spinal cord, and occasionally cerebral hemispheres. Well-circumscribed, not diffusely infiltrative.
Imaging
- Classic appearance: cystic mass with an enhancing mural nodule in the cerebellar hemisphere. If solid, still well-circumscribed. Pilocytic astrocytomas enhance intensely (unlike low-grade fibrillary astrocytomas, which may not enhance).
Histopathology
- Biphasic architecture: compact, densely fibrillar areas alternating with loose microcystic areas.
- Bipolar cells with long, thin "hairlike" (piloid) processes - GFAP-positive.
- Pathognomonic features: Rosenthal fibers (eosinophilic corkscrew-shaped inclusions) and eosinophilic granular bodies (mulberry-like inclusions).
- Microvascular proliferation and necrosis do NOT imply a worse prognosis (unlike other astrocytomas).
- Genetics: BRAF fusions (most commonly KIAA1549::BRAF) or BRAF V600E point mutations - activating the MAPK pathway. No IDH mutations.
4. Ependymoma
Typical Age & Demographics
- Bimodal presentation. In the first two decades: near the fourth ventricle (constitutes 5-10% of primary brain tumors in this age group). In adults: spinal cord is the most common site; strongly associated with neurofibromatosis type 2 (NF2).
Location
- Children/young: posterior fossa, fourth ventricle floor (characteristically arises from the floor, can extend through the foramina of Luschka and Magendie - "plastic" ependymoma).
- Adults: spinal cord (most common), especially in NF2.
- Supratentorial ependymomas occur (sometimes without obvious ventricular connection) and are often defined by ZFTA::RELA fusion.
Imaging
- Solid, non-infiltrative masses (unlike diffuse gliomas). Grainger & Allison describe them as well-defined.
- Posterior fossa tumors are hypointense to isointense on T1, hyperintense on T2, with heterogeneous enhancement.
- A nearly pathognomonic radiologic sign is extension through the foramina of the fourth ventricle ("plastic ependymoma").
Histopathology
- Solid, non-infiltrative growth pattern.
- Characteristic structures:
- Perivascular pseudorosettes: tumor cells radially arranged around blood vessels with a fibrillary anuclear zone between cells and vessel wall - the most consistent finding.
- True ependymal rosettes: tumor cells arranged around a central canal-like lumen (less common but more specific).
- Cells have round to oval nuclei with speckled ("salt-and-pepper") chromatin.
- Molecular subtypes (WHO 2021): posterior fossa A (PFA - poor prognosis, loss of H3K27me3) vs. posterior fossa B (PFB - better prognosis); supratentorial tumors often defined by ZFTA::RELA fusion (aggressive) or YAP1 fusion (favorable); spinal cord tumors: NF2 mutation (most common), or rare MYCN-amplified (poor prognosis).
- Spinal myxopapillary ependymoma: filum terminale, mucoid matrix, papillary architecture.
5. Oligodendroglioma
Typical Age & Demographics
- Account for 5-15% of gliomas. Most commonly detected in the fourth and fifth decades of life. Patients may have had several years of antecedent neurologic symptoms, often including seizures (high rate of epilepsy due to cortical involvement).
Location
- Cerebral hemispheres, predominantly frontal and temporal lobes. Diffusely infiltrative.
Imaging
- Infiltrative cortical/subcortical mass, often with cortical calcification (detectable on CT in up to 90% of cases - a key diagnostic clue).
- Low-grade tumors: T2/FLAIR hyperintense, typically non-enhancing. Higher-grade (anaplastic) tumors may enhance.
- "Bubbly" or heterogeneous appearance due to calcification, cyst formation, and focal hemorrhage.
Histopathology
- Infiltrative tumor with gelatinous gray cut surface; may show cysts, focal hemorrhage, and calcification.
- Pathognomonic appearance: sheets of uniform round cells with spherical nuclei and finely granular chromatin (similar to normal oligodendrocytes) surrounded by a clear cytoplasmic halo - the "fried egg" artifact (a fixation artifact).
- Delicate network of "chicken-wire" anastomosing capillaries branching around tumor cell groups.
- Calcification present in up to 90% of cases.
- Grade 2: low mitotic activity. Grade 3 (anaplastic): higher cellularity, nuclear atypia, increased mitoses, microvascular proliferation and/or necrosis.
- Genetics (required for diagnosis): IDH1 or IDH2 mutation + codeletion of chromosome 1p and 19q (1p/19q codeletion) - this combination is required for the WHO diagnosis. Most also have TERT promoter mutation (telomerase activation). ATRX is retained (unlike IDH-mutant astrocytoma).
- Prognosis is the best among diffuse gliomas: grade 2 - average survival 10-20 years; grade 3 - 5-10 years.
6. Glioblastoma (IDH-wildtype, WHO Grade 4)
Typical Age & Demographics
- Most common primary malignant brain tumor, accounting for ~14% of all primary CNS tumors and over half of all CNS malignancies. Predominantly occurs in the cerebral hemispheres of adults older than 55 years. Arises de novo (not from a prior low-grade lesion). Clinical course is rapidly progressive.
Location
- Cerebral hemispheres (most commonly frontal, temporal, parietal). Can cross the corpus callosum, producing a characteristic "butterfly glioma" pattern. Extremely rarely infratentorial.
Imaging
- Classic MRI appearance: ring-enhancing lesion with a central zone of necrosis surrounded by enhancing tumor rim, surrounded by T2/FLAIR hyperintensity (vasogenic edema + tumor infiltration).
- Marked heterogeneity: areas of necrosis (hypointense T1), hemorrhage, and hypervascularity.
- Contrast enhancement reflects disruption of the blood-brain barrier.
- Mass effect is pronounced.
Histopathology
- High variability from region to region - some areas gray-white and firm, others soft and yellow (necrosis) or red (hemorrhage and hypervascularity).
- Similar architecture to high-grade IDH-mutant astrocytoma but with greater necrosis and microvascular proliferation.
- Palisading necrosis: serpentine zones of necrosis bordered by tumor cells aligned perpendicularly - a defining pattern of glioblastoma.
- Microvascular proliferation: tufts of pleomorphic endothelial cells piling up and bulging into vessel lumens ("glomeruloid bodies") - driven by VEGF secreted by hypoxic malignant astrocytes.
- Nuclear atypia, abundant mitoses.
- IHC: GFAP-positive; IDH1 R132H immunostain negative (wildtype).
- Genetics: IDH-wildtype by definition. Most common alterations: combined +7/-10 (gain chromosome 7, loss chromosome 10), TERT promoter mutation, EGFR amplification. Also: CDKN2A homozygous deletion (loss of p16/Rb pathway), PTEN loss. Even an adult diffuse astrocytoma without histologic high-grade features is classified as glioblastoma if IDH-wildtype with at least one of these three genetic alterations.
- MGMT promoter methylation: predicts responsiveness to alkylating chemotherapy (temozolomide); present in ~40% of cases and associated with improved survival.
- Prognosis: median survival ~15 months with maximal resection + radiation + temozolomide; 25% survive 2 years.
Summary Table
| Feature | Meningioma | Medulloblastoma | Astrocytoma (IDH-mut) | Pilocytic Astrocytoma | Ependymoma | Oligodendroglioma | Glioblastoma (IDH-wt) |
|---|---|---|---|---|---|---|---|
| Typical age | Adult (F>M) | Children | Young/mid adult (~38 yr) | Children/young adults | Children (4th V); Adults (spinal) | 4th-5th decade | >55 years |
| Location | Dura; parasagittal, convexity, sphenoid wing | Cerebellar vermis/midline | Cerebral hemispheres | Cerebellum, optic tract | 4th ventricle (kids); spinal cord (adults) | Frontal/temporal lobes | Cerebral hemispheres |
| WHO Grade | 1-3 | 4 | 2-4 | 1 | 2-3 (location-dependent) | 2-3 | 4 |
| Imaging hallmark | Dural base + dural tail; homogeneous enhancement | Posterior fossa mass; obstructs 4th ventricle | Non-enhancing (G2); ring-enhancing (G4) | Cyst + mural nodule; intense enhancement | Solid, 4th ventricle; extends through foramina | Cortical calcification; "bubbly" | Ring-enhancing; necrotic center |
| Key histology | Whorls + psammoma bodies | Small blue cells; Homer Wright rosettes | Infiltrative; hyperchromatic nuclei; fibrillar background | Bipolar hairlike cells; Rosenthal fibers; eosinophilic granular bodies | Perivascular pseudorosettes; true ependymal rosettes | "Fried egg" cells; chicken-wire capillaries; calcification | Palisading necrosis; microvascular proliferation (glomeruloid) |
| Key genetics | NF2/merlin loss; TRAF7, AKT1 | WNT, SHH, TP53 pathways | IDH1/2-mut + TP53-mut + ATRX-mut | BRAF fusion/V600E (no IDH) | NF2 (spinal); ZFTA::RELA (supratentorial); PFA vs PFB (posterior fossa) | IDH-mut + 1p/19q codeletion + TERT-mut | IDH-wt + +7/-10 + TERT-mut + EGFR-amp |
| Prognosis | Excellent (G1 curative resection) | Variable by subtype; WNT ~100%, Group 3 poor | Grade 2: >10 yr; Grade 4: 2-4 yr | Excellent; often curable | Variable by type; PFA worst | Grade 2: 10-20 yr; Grade 3: 5-10 yr | ~15 months median survival |
Sources:
- Robbins & Cotran Pathologic Basis of Disease, Chapter 28 (CNS Tumors)
- Robbins & Kumar Basic Pathology, Chapter 21
- Bradley and Daroff's Neurology in Clinical Practice, Chapter 74
- Grainger & Allison's Diagnostic Radiology, Chapters 49 & 55
- Quick Compendium of Clinical Pathology, 5th ed.
This is a shared conversation. Sign in to Orris to start your own chat.