Aml vs cll vs cml vs all leukemia differentiate conscise
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
I now have everything needed for a thorough, concise comparison. Here is the full differentiation:
Leukemia Differentiation: AML vs CLL vs CML vs ALL
Quick Framework
| Feature | AML | ALL | CML | CLL |
|---|---|---|---|---|
| Cell line | Myeloid | Lymphoid | Myeloid | Lymphoid |
| Course | Acute | Acute | Chronic | Chronic |
| Blasts/cells | Myeloblasts | Lymphoblasts | All myeloid stages | Mature small lymphocytes |
| Blast cutoff | ≥20% blasts in BM | ≥20% blasts in BM | <10% (chronic phase) | Not blast-based |
1. AML - Acute Myeloid Leukemia
Who: Any age; median ~65-70 years. Risk factors: prior chemo/radiation, myelodysplasia, Down syndrome, benzene.
Cell of origin: Myeloid progenitor (granulocyte, monocyte, erythroid, megakaryocyte line)
Presentation:
- Abrupt onset: fatigue, fever, bleeding (thrombocytopenia), infections
- Hepatosplenomegaly, gum hypertrophy (M4/M5 monocytic subtypes)
- DIC - especially in APL (M3, PML-RARA fusion)
Hallmark morphology: Myeloblasts with Auer rods (pink needle-like cytoplasmic inclusions) - pathognomonic for AML. Large nucleoli described as "punched out."
Key genetics:
- t(8;21) RUNX1-RUNX1T1 - favorable
- inv(16) CBFB-MYH11 - favorable
- t(15;17) PML-RARA - APL (responds to ATRA)
- NPM1, CEBPA mutations - favorable
- FLT3-ITD - poor prognosis
- Complex karyotype, TP53 - poor
Treatment: Intensive "7+3" chemotherapy (cytarabine + anthracycline); ATRA + arsenic for APL; allogeneic stem cell transplant for high-risk disease; targeted agents (FLT3 inhibitors - midostaurin, venetoclax + azacitidine for older patients)
2. ALL - Acute Lymphoblastic Leukemia
Who: Most common leukemia in children (peak 2-5 yrs); also seen in adults (worse prognosis in adults)
Cell of origin: Lymphoid precursor (B-cell ~75%, T-cell ~25%)
Presentation:
- Abrupt: fever, pallor, bleeding, bone pain (children)
- CNS involvement common - headache, cranial nerve palsies
- Mediastinal mass (T-ALL especially)
- Lymphadenopathy, hepatosplenomegaly
Hallmark morphology: Lymphoblasts - smaller than myeloblasts, high nuclear-to-cytoplasmic ratio, no Auer rods, less distinct nucleoli (see image A vs B above)
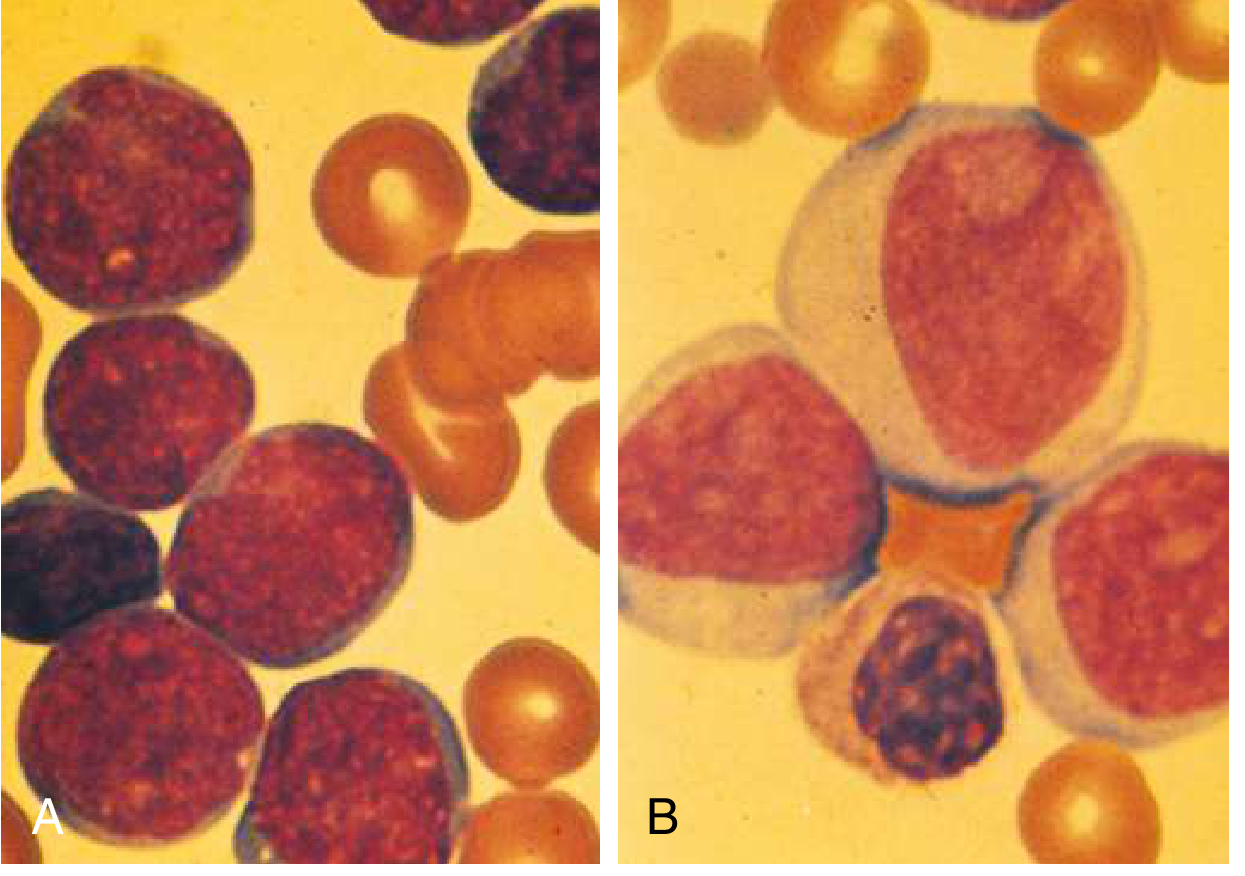
Key genetics:
- t(12;21) ETV6-RUNX1 - most common in children, best prognosis
- t(9;22) BCR-ABL1 (Ph+) - present in ~25% of adult ALL, worst prognosis (use TKI)
- t(v;11q23) KMT2A rearrangement - infant ALL, poor
- Hyperdiploidy (>50 chromosomes) - favorable (children)
- Hypodiploidy - unfavorable
Immunophenotype:
- B-ALL: CD19, CD22, CD10 (CALLA/CD10 positive = pre-B, best outcomes)
- T-ALL: CD7, CD3, CD1a
Treatment: Multi-drug induction (vincristine, prednisone, asparaginase ± anthracycline); CNS prophylaxis mandatory; TKI added for Ph+ ALL; maintenance therapy for 2-3 years; HSCT for high-risk
3. CML - Chronic Myeloid Leukemia
Who: Middle age (median ~50-65 yrs); slight male predominance
Cell of origin: Pluripotent myeloid stem cell
Presentation:
- Often insidious - found incidentally on CBC
- Massive splenomegaly (most prominent finding), fatigue, sweating, weight loss
- Basophilia characteristic
- Three phases: Chronic → Accelerated → Blast crisis
Hallmark genetics: t(9;22) - Philadelphia chromosome in >90% of cases
- BCR::ABL1 fusion gene → p210 oncoprotein → constitutive tyrosine kinase activity → uncontrolled myeloid proliferation
Lab findings:
- Leukocytosis (WBC often >50,000-100,000/µL) with full myeloid spectrum
- All stages of myeloid maturation on smear (left shift)
- Basophilia + eosinophilia
- Low LAP (leukocyte alkaline phosphatase) score - distinguishes from leukemoid reaction
- Thrombocytosis common
Phases:
- Chronic: <10% blasts, manageable, most patients at diagnosis
- Accelerated: 10-19% blasts, increasing basophils, refractory thrombocytopenia
- Blast crisis: ≥20% blasts (like acute leukemia), ~30% lymphoid, ~70% myeloid
Treatment: Tyrosine kinase inhibitors (TKIs) - first-line: imatinib, nilotinib, dasatinib, bosutinib; asciminib (STAMP mechanism, targets ABL1 myristoyl pocket) for resistant/T315I disease; allogeneic HSCT for blast crisis or TKI failure
4. CLL - Chronic Lymphocytic Leukemia
Who: Most common leukemia in adults in Western world (~15,000 new cases/year in US); median age 60 years; 2:1 male predominance; less common in Asia
Cell of origin: Mature B lymphocyte (CD5+ B cell)
Presentation:
- Often asymptomatic, found incidentally (absolute lymphocyte count >5,000/mm³)
- Lymphadenopathy, splenomegaly, hepatomegaly
- Recurrent infections (hypogammaglobulinemia)
- Autoimmune hemolytic anemia (AIHA), immune thrombocytopenia
- "B symptoms" in advanced disease: fever, night sweats, weight loss
Hallmark morphology:
- Small, mature-looking lymphocytes with scant cytoplasm
- Smudge cells (disrupted cells on smear) - pathognomonic
- Proliferation centers in lymph nodes (pathognomonic for CLL/SLL)
Key genetics (no translocations - mostly deletions):
- del(13q14) - most common, favorable (miR-15a/miR-16-1 loss → BCL2 overexpression)
- del(11q) - intermediate
- del(17p) / TP53 mutation - worst prognosis
- Trisomy 12q - intermediate
- Unmutated IGHV - aggressive; mutated IGHV - indolent
- NOTCH1 mutation (10-18%) - poor prognosis
Staging:
- Rai staging (0-IV) or Binet staging
- Stage 0 = lymphocytosis only; Stage IV = thrombocytopenia
Treatment: Many patients watch-and-wait initially. Treatment when symptomatic: BTK inhibitors (ibrutinib, acalabrutinib) - target B-cell receptor/BTK signaling; venetoclax + obinutuzumab (BCL2 inhibitor); FCR chemoimmunotherapy (now less common)
Side-by-Side Summary Table
| Feature | AML | ALL | CML | CLL |
|---|---|---|---|---|
| Age | Adults (elderly) | Children > adults | Middle age | Elderly (>60) |
| Onset | Acute (days-weeks) | Acute (days-weeks) | Insidious (months) | Insidious (years) |
| Key cell | Myeloblast | Lymphoblast | All myeloid stages | Mature B lymphocyte |
| WBC | Variable, often ↑ | Variable, often ↑ | Very high (>50K) | High lymphocytes |
| Spleen | Mild | Mild/moderate | Massive | Mild/moderate |
| Hallmark finding | Auer rods | No Auer rods, CNS involvement | Philadelphia chr., basophilia | Smudge cells |
| Key mutation | FLT3, NPM1, PML-RARA | BCR-ABL1 (Ph+), ETV6-RUNX1 | BCR-ABL1 (Ph+) | del(13q), IGHV status |
| Blast % | ≥20% | ≥20% | <10% (chronic) | Not applicable |
| Immunophenotype | CD13, CD33, CD117, MPO+ | CD19/22 (B) or CD3/7 (T) | CD13, CD33 (myeloid) | CD5+, CD19+, CD23+ |
| DIC risk | Yes (esp. APL) | Low | Low | Low |
| CNS involvement | Rare | Common | Rare | Rare |
| Prognosis | Variable; poor in elderly | Children: excellent; adults: moderate | Excellent with TKIs | Variable; often indolent |
| 1st-line Rx | 7+3 chemo (ATRA for APL) | Multi-agent + CNS prophylaxis | Imatinib/TKI | Watch-and-wait; ibrutinib/venetoclax |
Key Memory Aids
- AML = Auer rods, adults, myeloblasts, 7+3
- ALL = children, lymphoblasts, CNS, Ph+ = worst, CALLA+ = best
- CML = Philadelphia chromosome, massive spleen, TKIs cure-like response, three phases
- CLL = smudge cells, elderly, CD5+ B cells, most indolent, BTK inhibitors
Sources: Goldman-Cecil Medicine, Harrison's Principles of Internal Medicine 22E (2025), Robbins & Cotran Pathologic Basis of Disease
This is a shared conversation. Sign in to Orris to start your own chat.