Explain inflammmation
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
I now have rich, detailed content from the textbooks with excellent images. I have enough to write a comprehensive, well-structured answer on inflammation. Let me compile everything.
Inflammation
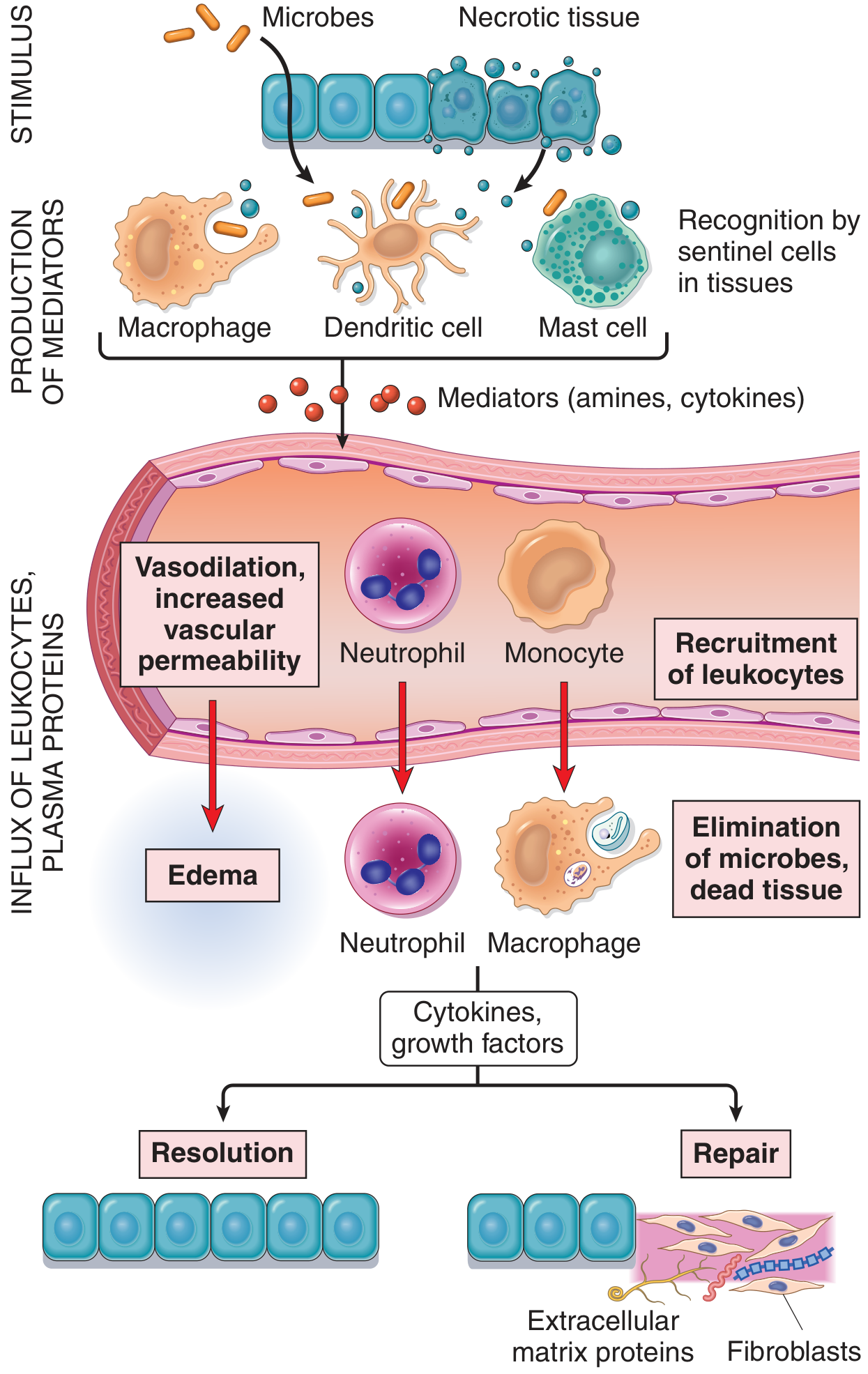
Inflammation is the response of vascularized tissues to infections and tissue damage that delivers leukocytes and molecules of host defense from the circulation to the site where they are needed, to eliminate offending agents and initiate repair. Despite its negative connotation in everyday language, it is fundamentally a protective process - without it, infections would go unchecked and wounds would never heal.
The suffix -itis appended to an organ name always denotes inflammation there (appendicitis, meningitis, conjunctivitis, etc.).
Robbins & Kumar Basic Pathology, p. 43
Sequence of Events (The Five R's)
The typical inflammatory reaction progresses through five steps:
- Recognition - Tissue-resident sentinel cells (macrophages, dendritic cells, mast cells) detect microbial products or damage-associated molecular patterns (DAMPs) via pattern-recognition receptors (e.g., Toll-like receptors).
- Recruitment - Mediators are released, triggering vascular and cellular changes that bring circulating leukocytes and plasma proteins to the site.
- Removal - Phagocytic cells (neutrophils, macrophages) ingest and destroy microbes and dead cells.
- Regulation - Anti-inflammatory mechanisms terminate the reaction once its purpose is accomplished.
- Repair - Damaged tissue is healed by regeneration of surviving cells or replacement with connective tissue (scarring).
Cardinal Signs
The external manifestations of inflammation were identified over 2,000 years ago:
| Latin | English | Cause |
|---|---|---|
| Calor | Heat | Increased blood flow |
| Rubor | Redness | Vasodilation |
| Tumor | Swelling | Edema (vascular leakage) |
| Dolor | Pain | Bradykinin, prostaglandins, substance P |
| Functio laesa | Loss of function | Added by Rudolf Virchow in the 19th century |
Acute vs. Chronic Inflammation
| Feature | Acute Inflammation | Chronic Inflammation |
|---|---|---|
| Onset | Fast: minutes to hours | Slow: days |
| Cellular infiltrate | Mainly neutrophils | Monocytes/macrophages and lymphocytes |
| Tissue injury | Usually mild and self-limited | May be significant |
| Fibrosis | None | May be severe and progressive |
| Local/systemic signs | Prominent | Variable, usually modest |
Robbins & Kumar Basic Pathology, Table 2.1
Acute Inflammation
Vascular Changes
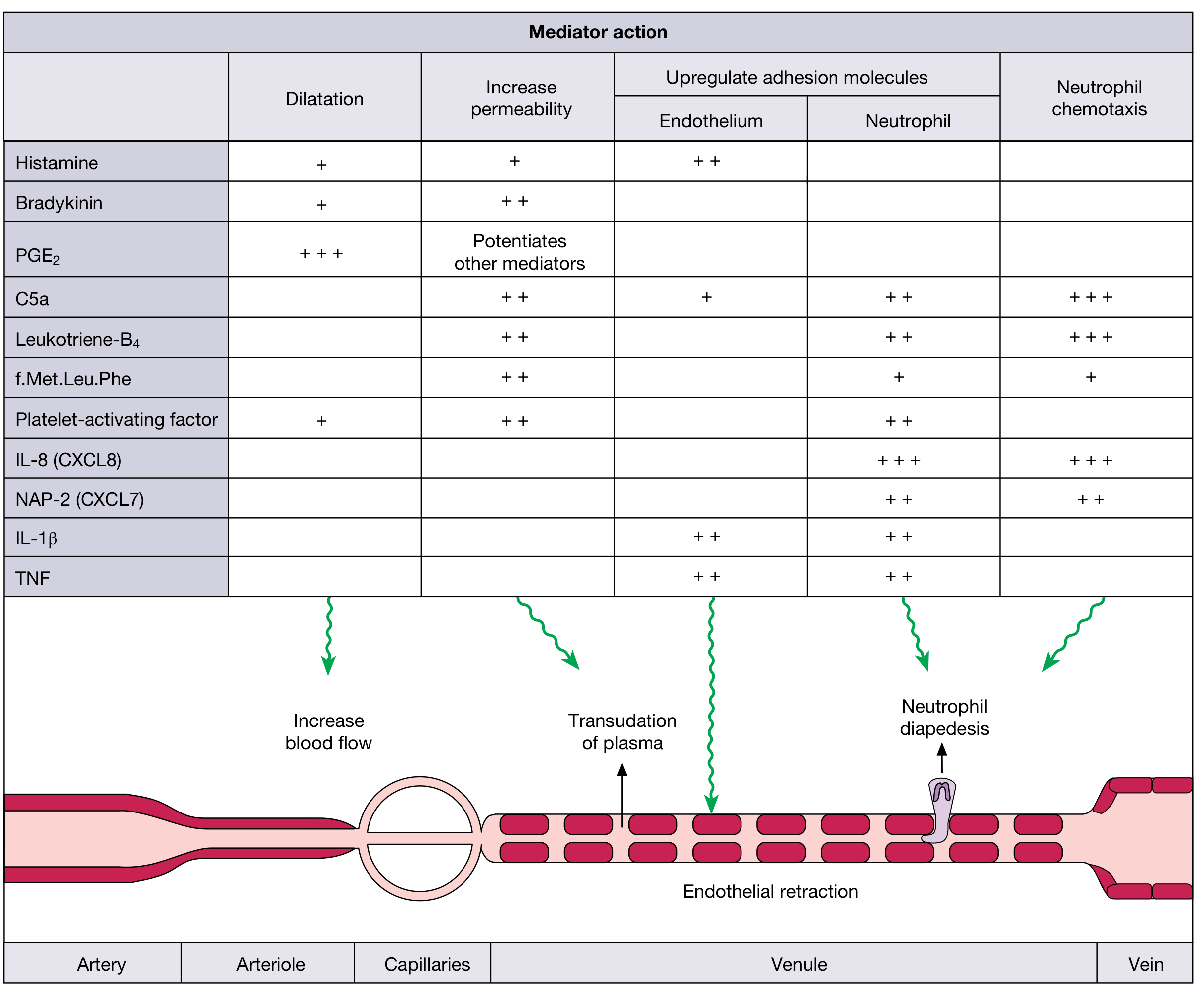
The first vascular event is vasodilatation (primarily of arterioles), which increases blood flow, causing heat and redness. This is followed by increased vascular permeability - the leakage of fluid and plasma proteins from venules into the interstitium. This protein-rich fluid (exudate) causes edema (swelling). The mechanisms include:
- Endothelial contraction forming intercellular gaps (histamine, bradykinin, leukotrienes)
- Direct endothelial injury
- Leukocyte-mediated endothelial damage
Leukocyte Recruitment
Leukocytes must travel from the bloodstream to the site of injury through a coordinated multi-step process:
- Margination and rolling - Due to slowed blood flow, leukocytes move to the vessel periphery; selectin molecules mediate loose rolling along the endothelium.
- Adhesion - Firm adhesion is mediated by integrins on leukocytes binding to ICAM-1 on endothelium, both upregulated by TNF and IL-1.
- Transmigration (diapedesis) - Leukocytes squeeze through endothelial junctions using PECAM-1 (CD31).
- Chemotaxis - Leukocytes migrate along a chemical gradient toward the site of injury. Major chemotactic agents include: bacterial products, complement fragment C5a, leukotriene B4 (LTB4), and chemokines (especially IL-8/CXCL8).
Neutrophils arrive first (within minutes to hours) due to their abundance in blood and rapid response. Monocytes follow within 24-48 hours and differentiate into macrophages with longer-lasting activity.
Phagocytosis
Once at the site, phagocytes ingest microbes and debris. The process is enhanced by opsonins (antibodies and C3b complement) that coat the target and are recognized by phagocyte receptors. The particle is engulfed into a phagosome, which fuses with lysosomes to form a phagolysosome, where killing occurs.
Killing mechanisms include:
- Reactive oxygen species (ROS) - generated by NADPH oxidase ("respiratory burst"); myeloperoxidase (MPO) converts H₂O₂ + Cl⁻ into hypochlorite, the most potent bactericidal agent in neutrophils.
- Nitric oxide (NO) - produced by iNOS in macrophages; reacts with O₂⁻ to form peroxynitrite (ONOO⁻), a highly reactive free radical.
- Lysosomal enzymes - acid hydrolases, elastase, cathepsins, defensins.
- Neutrophil extracellular traps (NETs) - extracellular webs of nuclear material and antimicrobial proteins that trap and kill extracellular pathogens.
Mediators of Inflammation
Mediators are either cell-derived (released from granules or synthesized de novo) or plasma-derived (circulating inactive precursors activated at the site). The major ones:
1. Vasoactive Amines
- Histamine - stored in mast cell granules; rapidly released in response to IgE crosslinking, C3a/C5a, or physical injury; causes arteriolar dilation and increased venular permeability via H1 receptors.
- Serotonin - present in platelets and neuroendocrine cells; similar effects to histamine.
2. Arachidonic Acid Metabolites
Generated from cell membrane phospholipids by phospholipase A2:
- Prostaglandins (via COX pathway) - PGE2 and PGI2 cause vasodilation, potentiate permeability, and cause pain and fever. NSAIDs (aspirin, ibuprofen) inhibit COX and thus block prostaglandin synthesis.
- Leukotrienes (via 5-LOX pathway) - LTB4 is a potent chemoattractant for neutrophils; LTC4, LTD4, LTE4 cause vasoconstriction, bronchoconstriction, and increased permeability. These are important in asthma. Montelukast blocks leukotriene receptors.
3. Cytokines and Chemokines
- TNF and IL-1 - produced by macrophages, dendritic cells, and mast cells; stimulate endothelial adhesion molecule expression, fever (via hypothalamic prostaglandins), production of acute-phase proteins by the liver, and leukocyte mobilization from bone marrow. Together these account for most systemic effects of inflammation.
- IL-6 - stimulates hepatic acute-phase protein production.
- Chemokines - a family of ~40 small proteins that guide leukocyte migration; IL-8 (CXCL8) is a major chemoattractant for neutrophils.
4. Complement System
The complement cascade (classical, lectin, and alternative pathways) generates:
- C3a and C5a (anaphylatoxins) - increase vascular permeability, cause mast cell degranulation.
- C5a - potent chemotactic agent for neutrophils.
- C3b - opsonin.
- Membrane attack complex (MAC, C5b-9) - directly lyses microbes.
5. Other Mediators
- Bradykinin - derived from kininogens by kallikrein; increases vascular permeability, causes smooth muscle contraction, vasodilation, and pain.
- PAF (platelet-activating factor) - phospholipid mediator; causes platelet aggregation, vasoconstriction, bronchoconstriction.
- Neuropeptides (substance P) - transmit pain signals and increase vascular permeability.
Morphologic Patterns of Acute Inflammation
| Pattern | Description | Example |
|---|---|---|
| Serous | Watery, protein-poor fluid accumulation | Blister after a burn |
| Fibrinous | Fibrin exudate due to severe vascular leakage | Fibrinous pericarditis ("bread and butter" appearance) |
| Purulent/Suppurative | Pus - neutrophils, necrotic cells, edema fluid | Abscess, furuncle |
| Ulcer | Local defect of epithelial surface | Peptic ulcer |
Outcomes of Acute Inflammation
Acute inflammation can resolve in three ways:
- Complete resolution - the ideal outcome; debris is cleared by macrophages, edema reabsorbed by lymphatics, tissue regenerates. Occurs when injury is limited and parenchymal cells can regenerate.
- Healing by scarring (fibrosis) - occurs when tissue destruction is extensive or the tissue cannot regenerate; connective tissue replaces the defect.
- Progression to chronic inflammation - occurs when the stimulus persists or healing is impaired.
Chronic Inflammation
Chronic inflammation is prolonged inflammation with ongoing tissue destruction and fibrosis occurring simultaneously with attempts at healing. It is characterized by:
- Infiltration by mononuclear cells - macrophages, lymphocytes, plasma cells.
- Tissue destruction by the inflammatory cells.
- Fibrosis and angiogenesis - attempts at repair.
Causes
- Persistent infections (e.g., M. tuberculosis, treponema, certain fungi) - organisms resist eradication.
- Autoimmune diseases (e.g., rheumatoid arthritis, lupus) - self-antigens drive the immune response.
- Prolonged exposure to toxic agents (e.g., silica causing silicosis, atherosclerosis from lipid deposition).
Key Cells
Macrophages are the dominant cells. They have two activation states:
- Classical (M1) macrophages - induced by IFN-γ and microbial products; produce ROS, NO, pro-inflammatory cytokines (TNF, IL-12); excellent at killing microbes.
- Alternative (M2) macrophages - induced by IL-4 and IL-13; primarily promote tissue repair, secrete growth factors, stimulate fibroblasts, synthesize collagen.
Lymphocytes (especially CD4+ T helper cells) interact with macrophages bidirectionally via cytokines:
- T cells produce IFN-γ → activates macrophages (M1)
- Macrophages present antigen and produce IL-12 → activate T cells
This bidirectional positive-feedback loop can perpetuate chronic inflammation.
Granulomatous Inflammation
A specific pattern of chronic inflammation featuring granulomas - focal accumulations of epithelioid macrophages (activated macrophages with abundant pink cytoplasm), often surrounded by lymphocytes, and sometimes containing multinucleated giant cells (fusion of macrophages).
Causes include:
- TB (Mycobacterium tuberculosis) - with central caseous necrosis
- Sarcoidosis - non-caseating granulomas
- Crohn's disease
- Fungal infections (histoplasma, cryptococcus)
- Foreign body reactions
Systemic Effects of Inflammation (Acute-Phase Response)
When inflammation is significant, systemic effects emerge, collectively called the acute-phase response, largely driven by TNF, IL-1, and IL-6:
| Effect | Mediator | Mechanism |
|---|---|---|
| Fever | TNF, IL-1, IL-6 | Stimulate PGE2 production in hypothalamus, raising the temperature set point |
| Acute-phase proteins | IL-6 | Liver produces CRP, fibrinogen, serum amyloid A; CRP acts as opsonin |
| Leukocytosis | TNF, IL-1 | Mobilize neutrophils from bone marrow; shift toward immature forms ("left shift") |
| Elevated ESR | Fibrinogen | Fibrinogen causes RBC rouleaux formation, accelerating sedimentation |
| Reduced appetite, sleep changes | TNF, IL-1 | Central effects |
In severe systemic infections, massive cytokine release can cause septic shock (hypotension, DIC, multi-organ failure) - an extreme and life-threatening consequence of systemic inflammation.
Summary
| Aspect | Key Points |
|---|---|
| Purpose | Protective; eliminates pathogens and initiates repair |
| Acute | Fast, neutrophil-dominated, vascular changes prominent |
| Chronic | Slow, macrophage/lymphocyte-dominated, fibrosis |
| Key mediators | Histamine, prostaglandins, leukotrienes, cytokines (TNF, IL-1), complement |
| Cardinal signs | Heat, redness, swelling, pain, loss of function |
| Outcomes | Resolution, scarring, or chronic inflammation |
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease; Robbins & Kumar Basic Pathology; Roitt's Essential Immunology
This is a shared conversation. Sign in to Orris to start your own chat.