Etiopathogeneamsis if RHD
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Here is a detailed, well-sourced account of the etiopathogenesis of Rheumatic Heart Disease (RHD).
Etiopathogenesis of Rheumatic Heart Disease (RHD)
RHD is the cardiac manifestation of Acute Rheumatic Fever (ARF), itself an immunologically mediated complication of Group A streptococcal (GAS) pharyngeal infection. It is fundamentally a disease of three interacting factors: the agent (GAS), the susceptible host, and an adverse environment.
1. The Agent - Group A Streptococcus (GAS)
- ARF follows pharyngeal infection with GAS (Group A beta-hemolytic Streptococcus pyogenes), not skin infection in most populations (though GAS skin infections may contribute in Africa via scabies-related pyoderma).
- The attack rate from untreated epidemic GAS pharyngitis is 1-3%, indicating that only genetically susceptible individuals develop ARF.
- The pathogenic potential resides in specific streptococcal antigens, particularly the M protein (an alpha-helical coiled-coil cell surface protein). Certain M protein serotypes (rheumatogenic strains) are especially likely to trigger the cross-reactive immune response.
- The group A carbohydrate (specifically N-acetyl-beta-D-glucosamine, GlcNAc) is another key antigen.
2. Molecular Mimicry - The Core Mechanism
The central pathogenetic mechanism is molecular mimicry: GAS antigens share structural/epitopic similarity with host cardiac proteins, causing the immune response to attack the heart.
| GAS Antigen | Cross-reacts with Host Protein |
|---|---|
| M protein (especially M5, M6) | Cardiac myosin (light meromyosin, LMM) |
| M protein | Tropomyosin, vimentin |
| N-acetyl-glucosamine (GlcNAc) | Cardiac myosin, laminin (valve basement membrane) |
| M protein epitopes | Neuronal antigens (caudate nucleus) - causes chorea |
The key epitope on M5/M6 protein is a five-amino-acid sequence Gln-Lys-Ser-Lys-Gln that cross-reacts with cardiac myosin. Up to 63.2% of intralesional T cell clones from RHD valve tissue react against cardiac light meromyosin peptides. - Firestein & Kelley's Textbook of Rheumatology
3. The Immune Response - Step by Step
Step 1: Innate immunity activation
GAS pharyngitis triggers neutrophils, macrophages, and dendritic cells to phagocytose bacteria and present antigens to T cells.
Step 2: Adaptive immune response
- Humoral: Cross-reactive antibodies (anti-M protein, anti-GlcNAc) are generated. These antibodies bind to valve endothelial cells and basement membrane (cross-reacting with laminin), generating inflammatory signals.
- Cellular: CD4+ T cells are activated. Intralesional T cells in valve tissue cross-react with streptococcal M5 peptides AND cardiac myosin, confirming dual specificity.
Step 3: The "Two-Hit" Hypothesis for valve damage
- First hit: Anti-GlcNAc and anti-myosin antibodies bind valve endothelium → upregulate VCAM-1 (vascular cell adhesion molecule-1) on the endothelial surface.
- Second hit: Upregulated VCAM-1 interacts with VLA-4 (very late activation antigen-4) on CD4+ T lymphocytes → T cells extravasate into valve tissue.
This results in a cytokine-rich, T cell-driven chronic valvulitis.
Step 4: Cytokine-mediated amplification
Four T-helper subsets (Th1, Th2, Th17, Tregs) participate:
- TNF-alpha, IFN-gamma (Th1) - drive pro-inflammatory macrophage activation
- IL-4, IL-10 (Th2) - modulate humoral response
- IL-17, IL-23 (Th17) - promote neutrophil recruitment and tissue damage
Key chemokines recruited to valve tissue include CCL1/I-309 and CXCL9/Mig, which mediate both CD4+ and CD8+ T cell infiltration. Matrix metalloproteinase-25 (MMP-25) is implicated in extracellular matrix degradation in the valve.
4. The Host - Genetic Susceptibility
- Only ~3-6% of individuals exposed to rheumatogenic GAS infection develop RF, suggesting genetic susceptibility is essential.
- Heritability of RF is ~60%.
- Individuals with a family history of RF have ~5x higher risk; children raised separately from parents with RHD still have a relative risk of 2.93.
- Genetic associations found in:
- HLA class II alleles (controlling adaptive immune response)
- Innate immunity genes: ficolin-2, mannose-binding lectin-2, TLR2
- Cytokine genes: TNF-alpha, TGF-beta, IL-1RA, IL-10
- B-cell alloantigens (D8/17 antigen historically associated)
- Women are approximately 1.8x more susceptible to developing RHD (also influenced by pregnancy exposure and child-rearing proximity to GAS).
5. The Environment
RF is a disease of poverty:
- Associated with overcrowding, poor housing, malnutrition, low income, limited healthcare access.
- The incidence fell in industrialized countries before the antibiotic era, driven by improved socioeconomic conditions.
- In endemic regions (Sub-Saharan Africa, South Asia, Pacific Islands, indigenous Australian/New Zealand communities), incidence among 5-14 year-olds can reach 162-228/100,000/year.
6. Pathologic Features (Acute ARF → Chronic RHD)
Acute Phase - ARF causes Pancarditis
The pathology affects all three layers of the heart:
| Layer | Acute Finding |
|---|---|
| Pericardium | Fibrinous exudate (usually resolves without sequelae) |
| Myocardium | Aschoff bodies (pathognomonic) |
| Endocardium/Valves | Fibrinoid necrosis + 1-2mm fibrin vegetations (verrucae) along valve closure lines |
The Aschoff Body (Pathognomonic Lesion)
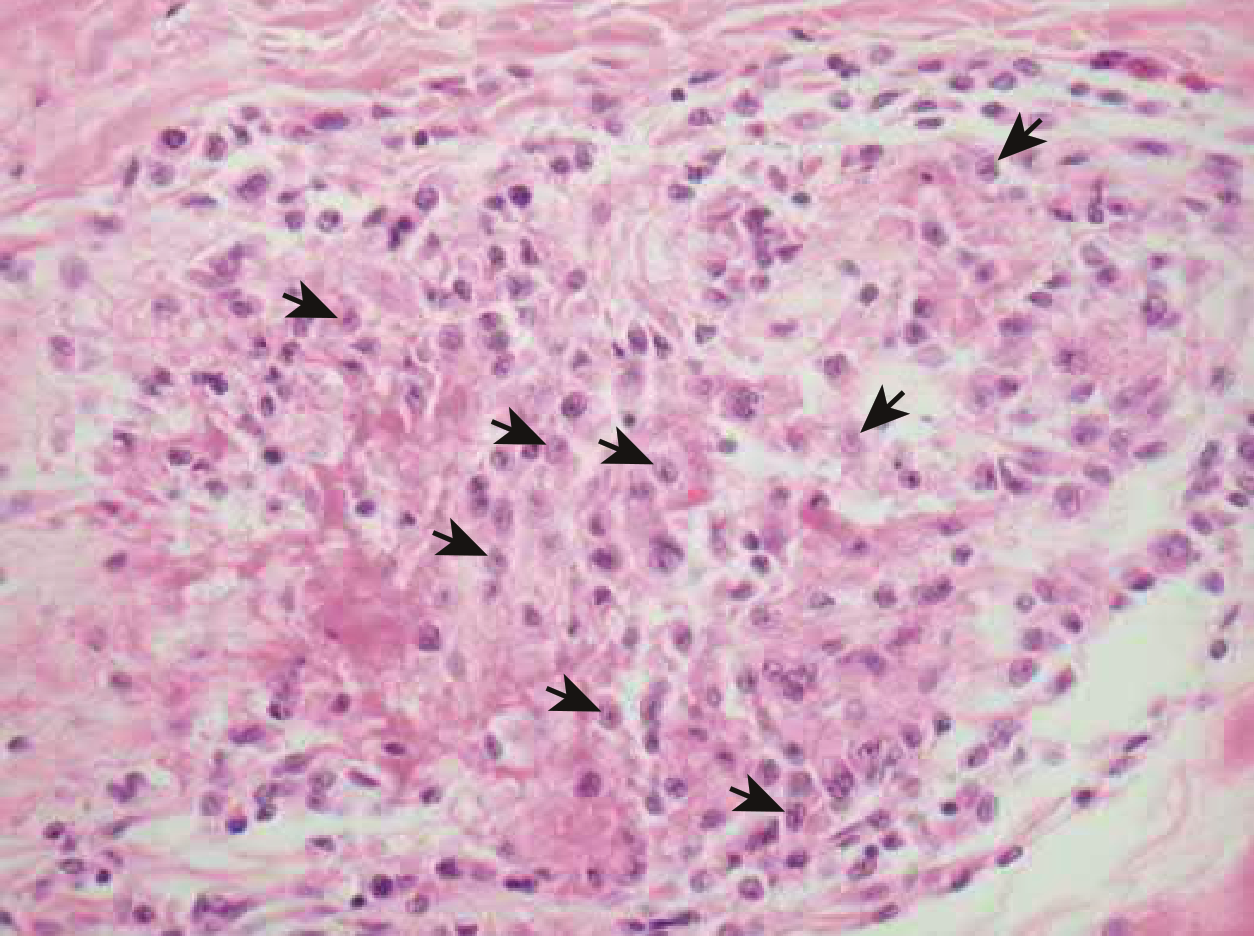
Aschoff body: composed of T lymphocytes, plasma cells, and plump activated macrophages called Anitschkow (caterpillar) cells - these have abundant cytoplasm and a central wavy chromatin ribbon resembling a caterpillar. Surrounded by a zone of fibrinoid necrosis. - Braunwald's Heart Disease, p. 856
Chronic Phase - Scarring and Valvular Distortion
Repeated episodes of ARF → cumulative valve damage → chronic RHD:
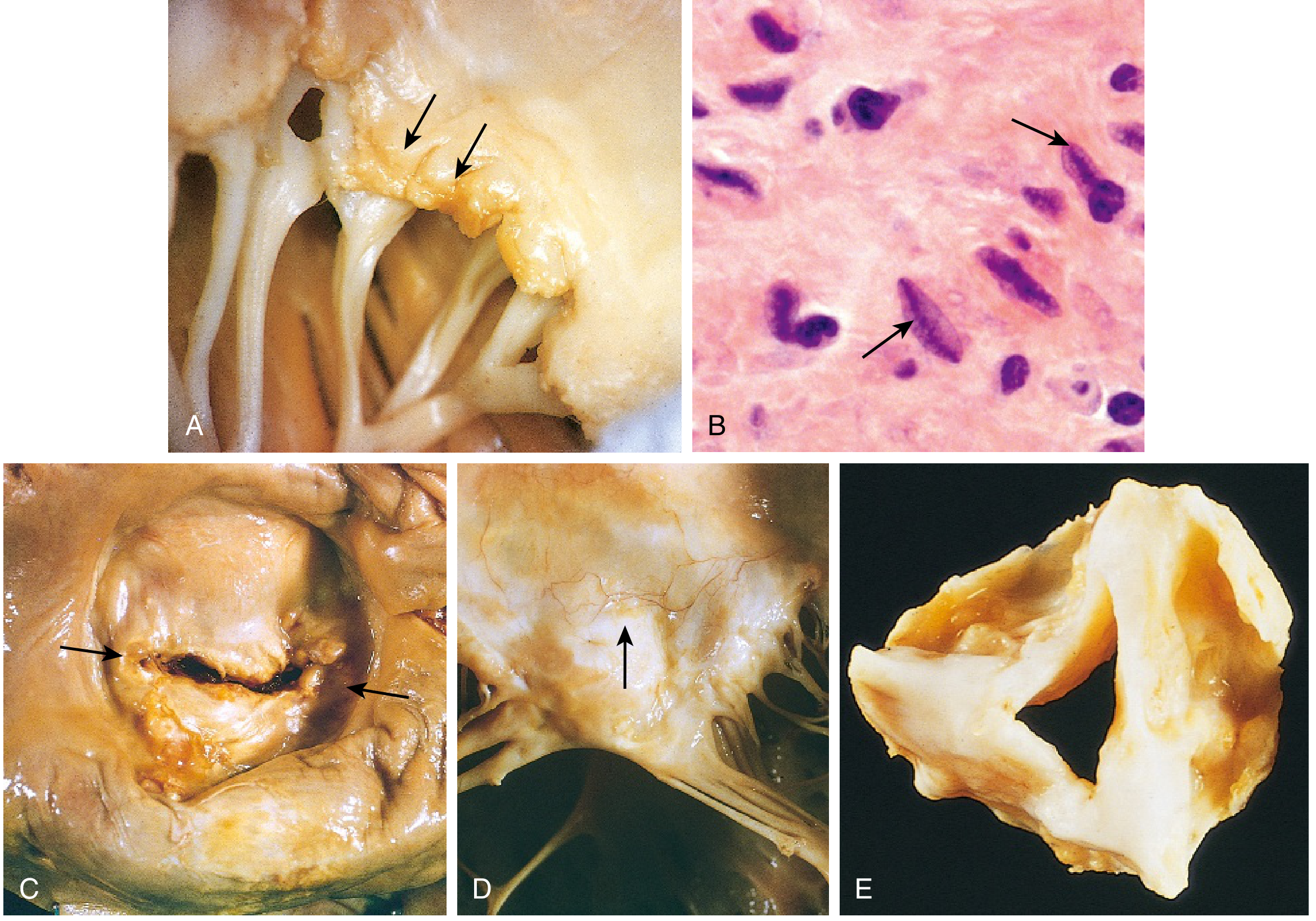
Fig 9.19 (Robbins & Kumar Basic Pathology): Acute rheumatic mitral valvulitis (A), Anitschkow cells (B), classic mitral stenosis with commissural fusion and chordal thickening (C-D), rheumatic aortic stenosis (E).
Key chronic changes:
- Aschoff bodies replaced by fibrous scar
- Valve leaflets thickened, retracted, fibrotic
- Commissural fusion ("fishmouth" / "buttonhole" stenoses)
- Thickening and fusion of chordae tendineae
- Neovascularization within leaflets
- Progressive calcification
7. Valve Distribution in Chronic RHD
| Valve Involved | Frequency |
|---|---|
| Mitral alone | 70% |
| Mitral + Aortic | ~25% |
| Tricuspid (less severely) | Less common |
| Pulmonic | Almost never |
Mitral stenosis is essentially the only acquired cause of isolated mitral stenosis. The left atrium dilates under pressure, leading to atrial fibrillation, mural thrombus formation, and eventually pulmonary hypertension and right heart failure. - Robbins & Kumar Basic Pathology, p. 366-367
Summary Diagram of Pathogenesis
GAS pharyngitis (rheumatogenic M-type)
↓
Immune activation (innate + adaptive)
↓
Cross-reactive antibodies & T cells (Molecular Mimicry)
- Anti-M protein ←→ cardiac myosin, tropomyosin
- Anti-GlcNAc ←→ laminin (valve basement membrane)
↓
"Two-hit": antibody → VCAM-1 upregulation → T cell infiltration
↓
Acute valvulitis (verrucae, Aschoff bodies)
↓
Repeated ARF episodes (untreated strep pharyngitis)
↓
Cumulative valve scarring → fibrosis → stenosis/regurgitation
↓
CHRONIC RHD
Sources: Braunwald's Heart Disease (p. 855-856), Robbins & Kumar Basic Pathology (p. 366-367), Firestein & Kelley's Textbook of Rheumatology (pp. 2578-2579), Harrison's Principles of Internal Medicine 22E.
This is a shared conversation. Sign in to Orris to start your own chat.