Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathophysiology
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Loading Image
Loading Image
Reading File
I now have comprehensive source material from Harrison's, Goldman-Cecil, Murray & Nadel, Robbins, and Sabiston. I have enough to write a thorough, well-structured answer.
Acute Respiratory Distress Syndrome (ARDS): Mechanism
Overview
ARDS is a clinical syndrome of severe hypoxemic respiratory failure caused by diffuse alveolar damage (DAD). It is defined by the Berlin criteria: bilateral pulmonary infiltrates, a PaO2/FiO2 (P/F) ratio <300 mmHg, onset within 7 days of a known insult, and respiratory failure not fully explained by cardiac failure or fluid overload (mild <300, moderate <200, severe <100).
Phases of ARDS
The condition progresses through three overlapping phases:

Figure: Time course of ARDS phases - Harrison's Principles of Internal Medicine 22E
Phase 1: Exudative Phase (Days 0-7)
This is the acute injury phase, characterized by loss of the alveolar-capillary barrier.
Triggering Insults
ARDS is initiated by direct (pulmonary) or indirect (extrapulmonary) insults:
- Direct: pneumonia, aspiration, inhalation injury
- Indirect: sepsis, severe trauma, pancreatitis, blood product transfusions, hemorrhage/hypotension
Step-by-step Pathogenesis
1. Initial insult and pattern recognition
Bacteria, viruses, or other danger signals activate Toll-like receptors (TLRs) on alveolar type I (ATI) epithelial cells and resident alveolar macrophages. This triggers secretion of chemokines (IL-8, leukotriene B4) that recruit circulating immune cells.
2. Neutrophil recruitment and activation - the central effector
Neutrophils are the primary mediators of endothelial and epithelial injury. They transmigrate across the alveolar-capillary membrane and release:
- Proteases (elastase, metalloproteinases) - digest the extracellular matrix and basement membrane
- Reactive oxygen species (ROS) - cause direct oxidative cell injury
- Neutrophil extracellular traps (NETs) - contribute to further epithelial and endothelial damage
- Activated platelets form aggregates with PMNs, amplifying the injury
3. Cytokine storm
Proinflammatory cytokines surge in the alveolar space:
- TNF-α, IL-1β, IL-6, IL-8 are markedly elevated
- These further recruit and activate neutrophils, establishing a self-amplifying inflammatory loop
- Monocytes also migrate into the lung and cause epithelial apoptosis via IFN-β-dependent release of TRAIL (TNF-related apoptosis-inducing ligand)
4. Alveolar-capillary barrier disruption
The combined effect of these mediators is loss of the normally tight alveolar barrier, affecting both layers:
- Endothelial injury: disrupted tight junctions → capillary leak → protein-rich fluid floods the interstitium
- Epithelial injury: type I pneumocytes (which cover ~95% of the alveolar surface) are necrotic and stripped; type II pneumocytes are also damaged
5. Alveolar flooding and surfactant dysfunction
- Protein-rich edema fluid accumulates in alveolar spaces
- The edema fluid inactivates pulmonary surfactant; damaged type II pneumocytes also fail to produce adequate surfactant
- In pancreatitis-associated ARDS, phospholipase A2 specifically degrades surfactant phospholipids, promoting alveolar collapse
- Condensed plasma proteins + cellular debris + dysfunctional surfactant aggregate to form the pathognomonic hyaline membranes (seen on histology as eosinophilic, whorled deposits lining the alveolar walls)
6. Impaired fluid clearance
Injury to Na+/K+-ATPase and epithelial sodium channels (ENaC) on type II pneumocytes impairs active fluid resorption from the alveolar space, perpetuating flooding.
7. Vascular injury and pulmonary hypertension
Microvascular injury occurs in parallel:
- Microthrombi form from platelet-PMN aggregates and fibrin deposition (procoagulant state with suppressed fibrinolysis)
- Fibrocellular proliferation obliterates small pulmonary vessels
- Endothelin and thromboxane cause vasoconstriction
- Result: pulmonary vascular resistance increases, dead space rises, and pulmonary hypertension develops
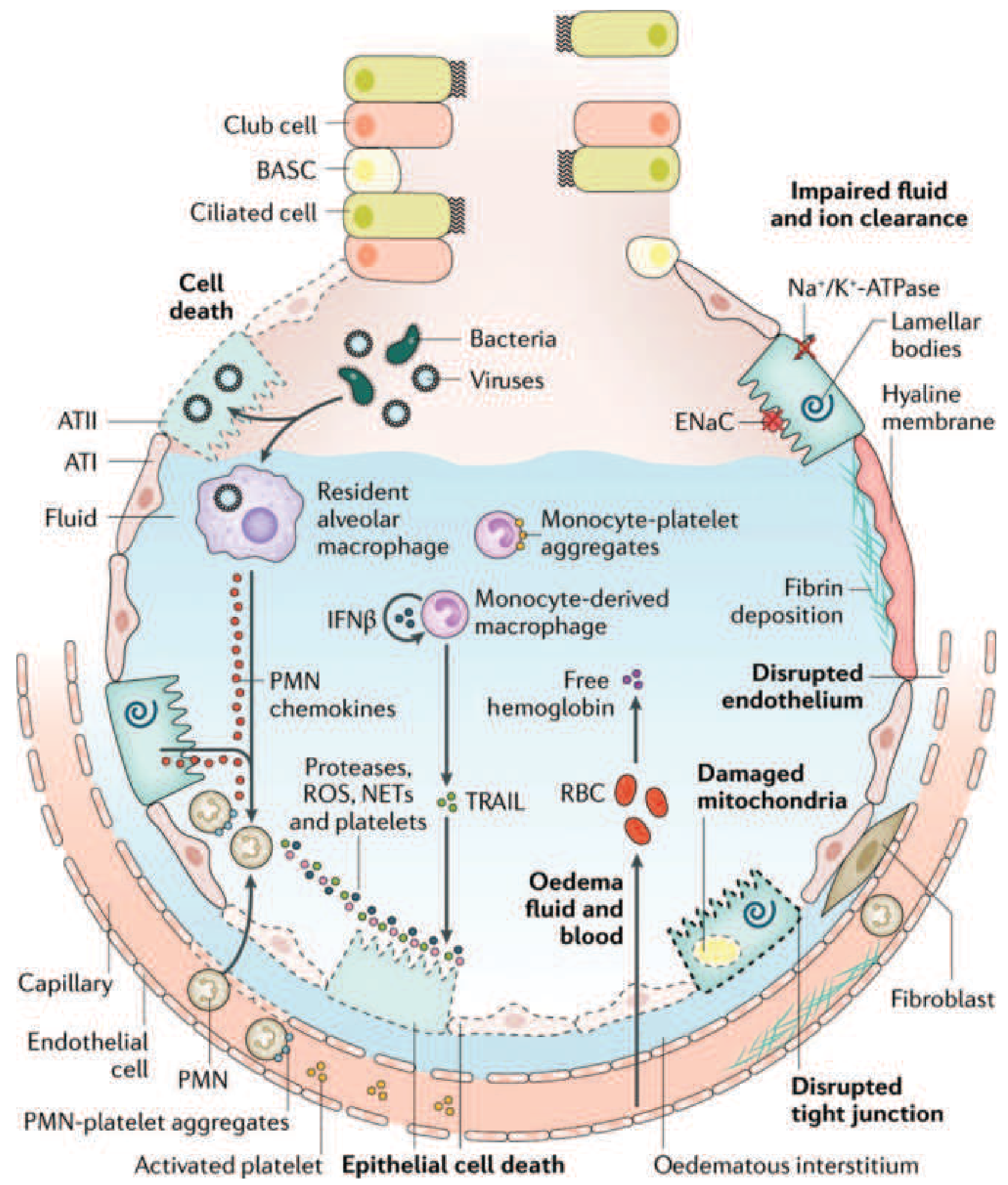
Figure 312-3: The injured alveolus in the acute phase of ARDS. Bacteria/viruses injure ATI epithelial cells; TLR activation drives macrophage chemokine release, neutrophil (PMN) transmigration, ROS/NETs/protease release, TRAIL-mediated apoptosis, fibrin deposition, and impaired ENaC/Na-K-ATPase fluid clearance - Harrison's 22E (adapted from Matthay et al., Nat Rev Dis Primers, 2019)
Physiological Consequences of the Exudative Phase
| Mechanism | Consequence |
|---|---|
| Alveolar flooding (dependent zones) | Intrapulmonary shunt → refractory hypoxemia |
| Surfactant loss → alveolar collapse | Reduced lung compliance, increased work of breathing |
| Microvascular obliteration | Increased dead space → hypercapnia |
| Pulmonary vasoconstriction + microthrombi | Pulmonary hypertension, RV strain |
| Hypoxemia + hypercapnia | Impaired ion transport → worsened fluid clearance |
The result is the characteristic presentation: tachypnea, progressive dyspnea, and refractory hypoxemia unresponsive to supplemental oxygen alone. Chest imaging shows bilateral opacities with a gravitational gradient - dependent zones are consolidated/fluid-filled while non-dependent zones may appear relatively spared (giving the "baby lung" concept that underpins lung-protective ventilation).
Phase 2: Proliferative Phase (Days 7-21)
Most patients begin recovering during this phase. Histological changes include:
- Shift from neutrophil-predominant to lymphocyte-predominant infiltrates
- Type II pneumocyte proliferation along alveolar basement membranes - these cells synthesize new surfactant and differentiate into type I pneumocytes, restoring the epithelial barrier
- Organization of alveolar exudates
- Initiation of lung repair
However, in some patients, the reparative process goes awry, with early fibrotic changes appearing in the interstitium and alveolar walls.
Phase 3: Fibrotic Phase (Day 21+)
A subset of patients enter this phase despite treatment. Key pathological features:
- Alveolar edema and inflammatory exudates are replaced by extensive alveolar-duct and interstitial fibrosis
- Disrupted acinar architecture creates emphysema-like changes with large bullae
- Intimal fibroproliferation causes progressive pulmonary vascular occlusion and worsening pulmonary hypertension
Physiological consequences include:
- Reduced lung compliance
- Increased dead space
- Risk of pneumothorax
- Need for long-term mechanical ventilation and/or supplemental oxygen
Lung biopsy evidence of pulmonary fibrosis at any phase is independently associated with increased mortality.
Role of Ventilator-Induced Lung Injury (VILI)
Mechanical ventilation, while life-saving, can propagate the injury via two mechanisms:
- Volutrauma/barotrauma: repeated alveolar overdistention from excess tidal volume
- Atelectrauma: recurrent collapse and re-expansion of unstable alveoli
Because ARDS causes heterogeneous lung involvement, tidal volumes that are safe in "normal" lung regions may overdistend those regions while trying to recruit consolidated zones. This is the rationale for low tidal volume (6 mL/kg predicted body weight) ventilation - the only ventilatory strategy with proven mortality benefit.
Summary: Key Pathogenetic Sequence
Insult (sepsis/trauma/pneumonia etc.)
↓
TLR activation on alveolar macrophages + ATI cells
↓
Cytokine release (TNF-α, IL-1β, IL-6, IL-8)
↓
Neutrophil recruitment & activation
↓
Proteases + ROS + NETs → epithelial & endothelial injury
↓
Barrier breakdown → protein-rich alveolar edema
↓
Surfactant inactivation + type II cell damage
↓
Hyaline membrane formation
↓
Alveolar flooding → shunt → hypoxemia
Microvascular obliteration → dead space → hypercapnia
↓
[If repair fails] → fibrosis → chronic respiratory failure
Sources:
- Harrison's Principles of Internal Medicine, 22E, Chapter 312 (ARDS - Clinical Course and Pathophysiology)
- Murray & Nadel's Textbook of Respiratory Medicine, 2-Volume, Chapter 134
- Goldman-Cecil Medicine, Chapter 90 (Pathobiology)
- Robbins & Kumar Basic Pathology, Acute Lung Injury section
- Sabiston Textbook of Surgery, 11E, Chapter 11
This is a shared conversation. Sign in to Orris to start your own chat.