Portocaval anastomosis sites
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Here is a complete, textbook-sourced overview of portocaval (portosystemic) anastomosis sites.
Portocaval (Portosystemic) Anastomosis Sites
The portal venous system connects to the systemic (caval) venous system at several points where the two circulations lie in close apposition. Under normal conditions, flow through these connections is minimal. In portal hypertension, portal pressure rises above ~8-10 mmHg, reverses flow through these channels, and causes them to dilate into varices.
Diagram
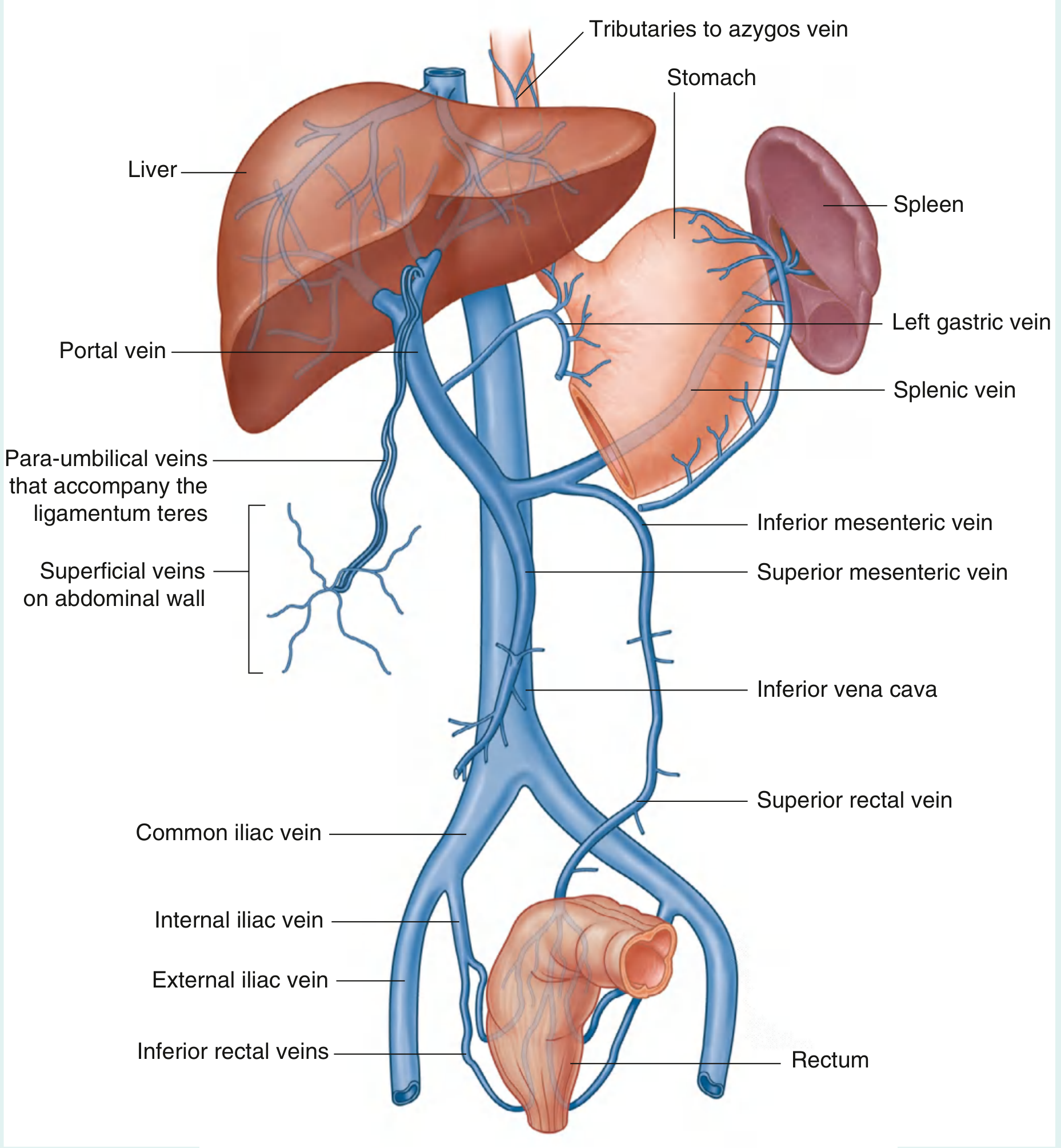
Fig. 4.134 - Portosystemic Anastomoses (Gray's Anatomy for Students)
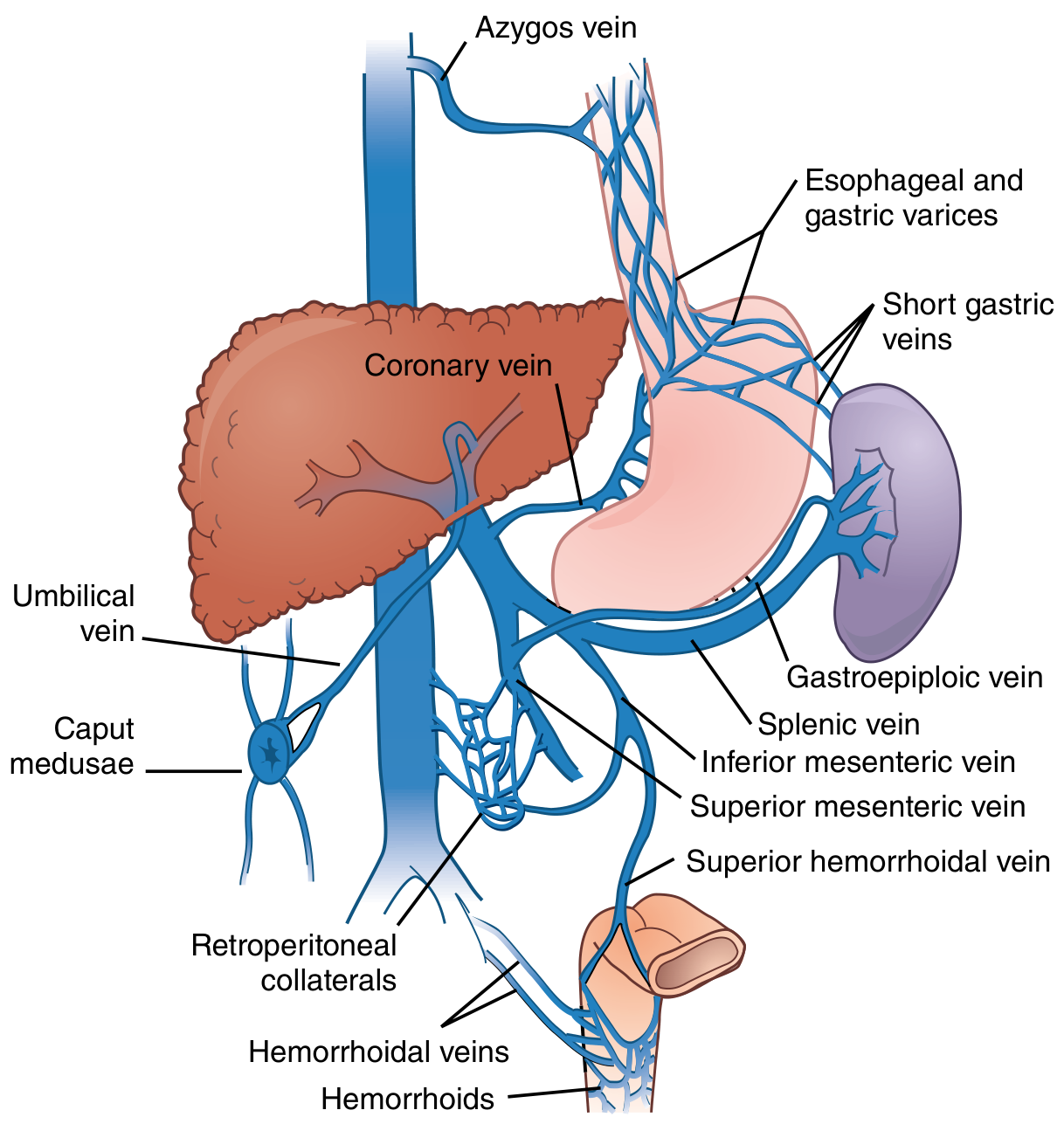
Portosystemic collateral pathways (Sabiston Textbook of Surgery)
The 4 Main Anastomotic Sites
1. Gastro-oesophageal Junction (Lower Oesophagus / Gastric Cardia)
| Portal tributary | Left gastric (coronary) vein + short gastric veins (vasa brevia) |
| Systemic tributary | Oesophageal veins → azygos vein → SVC |
| Varix formed | Oesophageal varices (± gastric varices) |
| Clinical significance | Most dangerous. Supplied mainly by the left gastric (coronary) vein. Oesophageal varices are prone to trauma and rupture, causing life-threatening haemorrhage. This is the clinically most important collateral. |
2. Umbilicus / Anterior Abdominal Wall
| Portal tributary | Para-umbilical veins (travel in ligamentum teres/round ligament of liver) → left branch of portal vein |
| Systemic tributary | Superficial epigastric veins, thoracoepigastric veins → external iliac / axillary veins |
| Varix formed | Caput medusae (dilated veins radiating from umbilicus) |
| Clinical significance | Caput medusae specifically indicates congestion of the left hepatic lobe because the para-umbilical veins drain into the left portal vein. This is the only site that communicates directly with the left branch of the portal vein; all others drain into the main portal trunk. |
3. Anorectal Junction (Lower Rectum / Anal Canal)
| Portal tributary | Superior rectal vein → inferior mesenteric vein |
| Systemic tributary | Middle and inferior rectal veins → internal iliac vein → IVC |
| Varix formed | Anorectal varices (not the same as primary haemorrhoids, though haemorrhoids may enlarge) |
| Clinical significance | Less dramatic than oesophageal varices but can bleed; important to distinguish true varices from haemorrhoids on examination. |
4. Retroperitoneal / Bare Area Collaterals (Veins of Retzius and Sappey)
| Portal tributaries | Intestinal, colic, and mesenteric veins; veins of the bare area of the liver |
| Systemic tributaries | Retroperitoneal veins draining to lumbar and renal veins → IVC |
| Varix formed | Retroperitoneal varices (clinically less visible) |
| Clinical significance | Identified during surgery; called veins of Retzius (retroperitoneal) and veins of Sappey (around the liver bare area/diaphragm). Can cause significant bleeding intraoperatively. |
Summary Table
| Site | Portal vein side | Systemic vein side | Clinical manifestation |
|---|---|---|---|
| Lower oesophagus / gastric cardia | Left gastric vein, short gastric veins | Azygos vein (via oesophageal veins) | Oesophageal/gastric varices |
| Umbilicus | Para-umbilical veins | Superficial epigastric, thoracoepigastric veins | Caput medusae |
| Anorectal junction | Superior rectal vein | Middle + inferior rectal veins | Anorectal varices |
| Retroperitoneum | Colic/mesenteric/intestinal veins | Retroperitoneal veins → IVC | Retroperitoneal varices (Retzius, Sappey) |
Key Clinical Points
- Portal hypertension threshold for collateral development is ~8-10 mmHg (HVPG). Clinically significant portal hypertension = HVPG >10 mmHg.
- The coronary (left gastric) vein is the main feeder of oesophagogastric varices and is the primary target in surgical shunt procedures.
- Caput medusae specifically indicates left portal vein hypertension; its presence points to left lobe congestion.
- In isolated splenic vein thrombosis (e.g., from pancreatitis), the left gastroepiploic vein becomes a major collateral and gastric (rather than oesophageal) varices predominate - this is "left-sided portal hypertension" and is cured by splenectomy alone.
- Intraoperative bleeding from retroperitoneal collaterals is a major hazard during abdominal surgery in cirrhotic patients.
Sources: Gray's Anatomy for Students, p. 419 | Sabiston Textbook of Surgery, p. 1877 | Schwartz's Principles of Surgery, Fig. 31-14 | Color Atlas of Human Anatomy, p. 558 | Bailey & Love's Short Practice of Surgery
Refer to standard pathology textbooks like Robbins medicine textbooks like Harrison Davidson to explain the pulmonary complications of liver failure and portal hypertension. Add disease identifiers
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Reading File
I now have comprehensive data from Robbins, Sleisenger & Fordtran, Murray & Nadel, and Fishman's. Let me compile the full answer.
Pulmonary Complications of Liver Failure and Portal Hypertension
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease | Sleisenger & Fordtran's Gastrointestinal and Liver Disease | Murray & Nadel's Textbook of Respiratory Medicine | Fishman's Pulmonary Diseases and Disorders
Robbins identifies two main pulmonary syndromes in the setting of chronic liver failure and portal hypertension, which are now expanded by respiratory medicine texts into four well-defined complications:
- Hepatopulmonary Syndrome (HPS)
- Portopulmonary Hypertension (POPH)
- Hepatic Hydrothorax
- General Pulmonary Function Disturbances
1. Hepatopulmonary Syndrome (HPS)
ICD-10: K76.81
Definition
HPS is defined by the clinical triad of:
- Intrapulmonary vasodilation (IPVD)
- Abnormal arterial oxygenation - alveolar-arterial PO₂ gradient (A-a)PO₂ ≥15 mmHg (or >20 mmHg if age >64 years) in the absence of another cause of hypoxemia
- Advanced liver disease (cirrhotic or non-cirrhotic portal hypertension)
(Murray & Nadel, per European Respiratory Society / International Liver Transplantation Society Task Force)
Epidemiology
- Occurs in 5-35% of patients with cirrhosis evaluated for liver transplantation
- Also described in non-cirrhotic portal hypertension and chronic hepatitis without confirmed cirrhosis
Pathogenesis
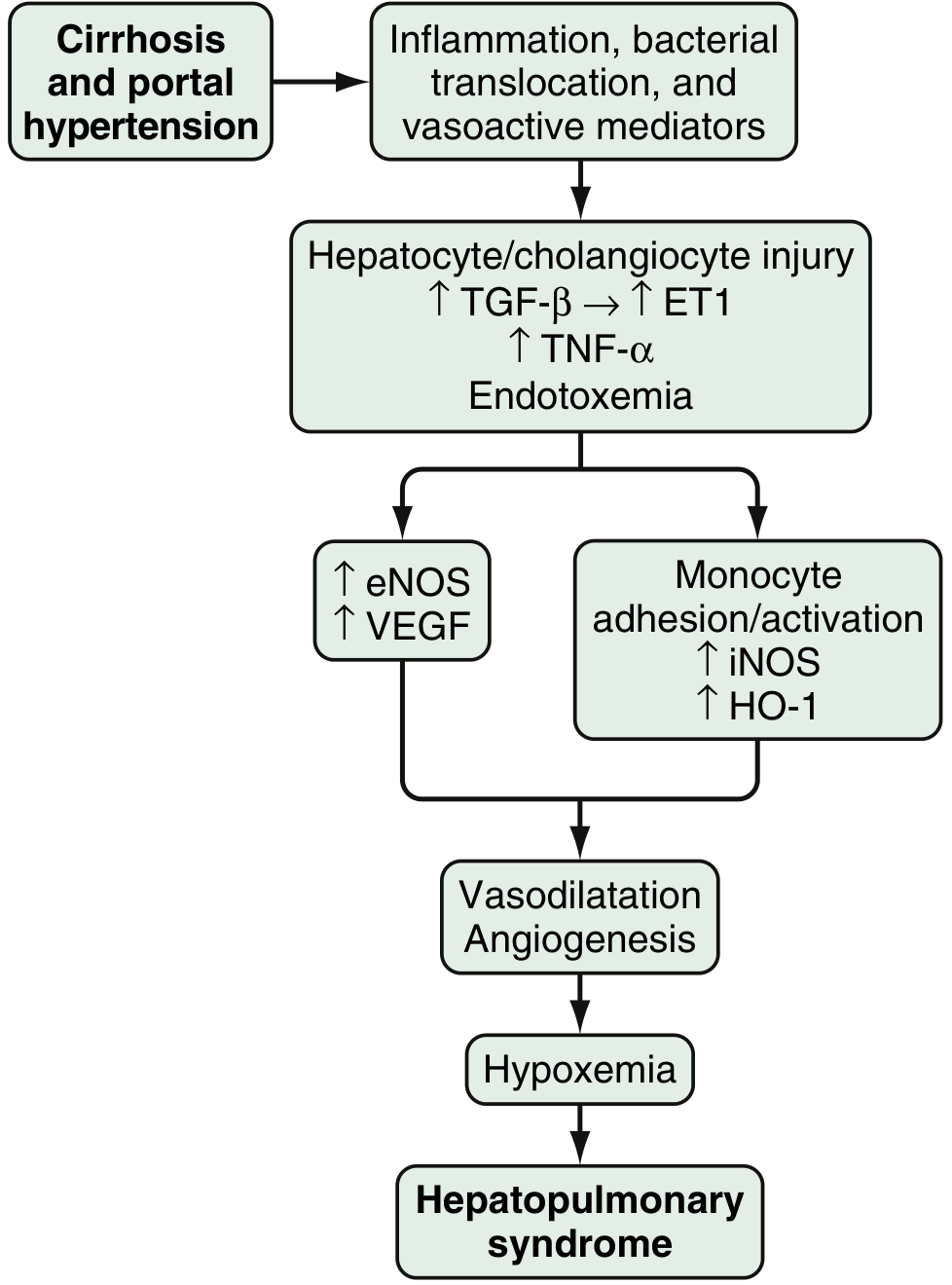
Fig. 94.3 - Proposed pathophysiology of HPS (Sleisenger & Fordtran's)
The core lesion is dilation of intrapulmonary precapillary and capillary vessels up to 500 μm in diameter (Robbins). This produces:
- Ventilation-perfusion (V/Q) mismatch - dilated vessels perfuse alveoli that are ventilated but cannot oxygenate blood efficiently
- Right-to-left shunting through dilated vessels
- Diffusion-perfusion impairment in severe cases - blood transits so quickly through widened capillaries that haemoglobin cannot equilibrate with alveolar O₂
Molecular mediators (Sleisenger & Fordtran):
- The injured liver releases endothelin-1 (ET-1), which paradoxically stimulates pulmonary microvascular endothelin-B receptors to upregulate eNOS (endothelial nitric oxide synthase) → excess nitric oxide (NO) production
- Monocyte adhesion in pulmonary vasculature activates iNOS, heme oxygenase-1 (HO-1, producing vasodilatory CO), and VEGF-mediated angiogenesis
- Gut bacterial translocation and endotoxemia drive the inflammatory cascade (TNF-α, TGF-β)
- The result: vasodilatation + angiogenesis → hypoxemia
Clinical Features
| Feature | Detail |
|---|---|
| Platypnea | Dyspnea worsened in erect posture, improved lying supine |
| Orthodeoxia | Hypoxemia worsened in upright position (gravity shifts blood to dependent, maximally dilated basal vessels) |
| Dyspnea | Insidious onset, slow progression |
| Digital clubbing | Seen alongside hypoxemia in advanced disease |
| Distal cyanosis | In severe HPS |
| Spider angiomata | Skin marker of underlying liver disease |
| Nocturnal desaturation | Occurs in up to 70% of HPS patients |
Grading of Severity (Task Force consensus)
| Grade | PaO₂ |
|---|---|
| Mild | ≥80 mmHg |
| Moderate | 60-80 mmHg |
| Severe | 50-60 mmHg |
| Very severe | <50 mmHg |
Diagnosis
- Contrast echocardiography (bubble echo): most sensitive test - microbubbles appear in the left-sided chambers after 3-5 cardiac cycles (late appearance = intrapulmonary shunting, vs. early appearance = intracardiac shunt). This confirms IPVD.
- ABG: elevated (A-a)PO₂; hypoxemia in moderate-severe cases
- CT chest: dilated peripheral pulmonary arteries in the lung bases
Treatment & Prognosis
- Definitive treatment: liver transplantation - resolves hypoxemia in most patients
- MELD exception points are allocated for advanced HPS to expedite transplantation
- Patients with pretransplant PaO₂ ≤44 mmHg have worse post-transplant survival
- No effective medical therapy; supplemental O₂ for palliation
2. Portopulmonary Hypertension (POPH)
ICD-10: I27.20 / K76.6
Definition
Pulmonary arterial hypertension (PAH) arising in the context of portal hypertension. By the Sixth World Symposium on Pulmonary Hypertension criteria (Group 1 PAH), POPH is defined hemodynamically as:
- Mean pulmonary arterial pressure (mPAP) >20 mmHg at rest
- Pulmonary artery wedge pressure (PAWP) ≤15 mmHg
- Pulmonary vascular resistance (PVR) ≥240 dynes·sec·cm⁻⁵ (normal <130)
Confirmed by right heart catheterization.
Epidemiology
- Prevalence: ~5-6% of patients evaluated for liver transplantation
- More common in women and patients with autoimmune hepatitis
- Hepatitis C is associated with a decreased risk
Pathogenesis (Robbins / Murray & Nadel)
- The mechanism is not fully understood. Unlike HPS (which causes vasodilation), POPH involves excessive pulmonary vasoconstriction and vascular remodeling.
- Portosystemic shunting allows vasoactive substances (thromboxanes, serotonin, neuropeptide Y, endothelin-1) that are normally cleared by the healthy liver to reach the pulmonary vasculature, causing vasoconstriction.
- Elevated macrophage migration inhibitory factor (proinflammatory), reduced bone morphogenetic protein 9 (endothelial quiescence factor), and altered estrogen metabolism have been implicated.
- Histopathology is identical to idiopathic PAH: intimal thickening, smooth muscle proliferation, plexogenic pulmonary arteriopathy, and in situ thrombosis - all contributing to elevated PVR. (Robbins)
Clinical Features
| Feature | Detail |
|---|---|
| Dyspnea on exertion | Most common symptom |
| Digital clubbing | Also seen in HPS |
| Right heart failure signs | JVD, loud P₂, tricuspid regurgitation murmur, lower limb oedema, abdominal distension |
| Syncope, chest pain | In advanced disease |
| Spirometry | Normal or near-normal (contrast with HPS where DLCO is reduced) |
| CXR | Enlarged cardiac silhouette, prominent pulmonary artery (in ~50-65%) |
Comparison: HPS vs. POPH
(Murray & Nadel, Table 126.3)
| Feature | HPS | POPH |
|---|---|---|
| Pathology | Precapillary/capillary vasodilation | Plexiform lesions, SMC proliferation, in situ thrombosis |
| Gas exchange | Markedly elevated (A-a)PO₂, moderate-severe hypoxemia | Normal or mild hypoxemia |
| Echo finding | Late microbubbles in left chambers | Elevated RVSP, RV dilation/dysfunction |
| Medical therapy | Supportive only | PAH-targeted therapy |
| Effect of liver Tx | Resolution of hypoxemia | Variable (50% remain on PAH therapy post-Tx) |
Treatment
- PAH-targeted vasodilator therapy: epoprostenol, treprostinil, sildenafil, tadalafil, bosentan, ambrisentan, macitentan (used similarly to idiopathic PAH)
- Calcium channel blockers: CONTRAINDICATED - worsen splanchnic vasodilation
- Beta-blockers: avoid if possible - worsen cardiac output and exercise capacity in POPH
- Liver transplantation: safe in selected patients with mPAP <35-45 mmHg and preserved RV function; liver Tx is contraindicated if mPAP >45-50 mmHg due to high perioperative mortality
Prognosis
- Without treatment: 5-year survival ~14%
- Modern PAH therapy era: 5-year survival ~40% (worse than idiopathic PAH despite better hemodynamics at baseline)
- Liver transplantation 1-year survival in selected POPH patients: 77-85%; highest risk in first 6 months
3. Hepatic Hydrothorax
ICD-10: J90 / K74.6
Definition
A transudative pleural effusion >500 mL in a patient with liver cirrhosis and portal hypertension, in the absence of primary cardiopulmonary disease. (Fishman's Pulmonary Diseases and Disorders)
Epidemiology
- Affects 5-10% of patients with cirrhosis
Pathophysiology
- Ascitic fluid passes from the peritoneal cavity into the pleural space through small diaphragmatic defects (<1 mm), usually in the tendinous portion of the right hemidiaphragm.
- The pressure gradient (positive intra-abdominal → negative intrapleural) drives unidirectional flow.
- Laterality:
- Right-sided: 80% (thinner right hemidiaphragm is more prone to defects)
- Left-sided: 18%
- Bilateral: 2%
- Up to 20% of patients have no detectable ascites (all ascitic fluid is decompressed into the chest), but residual ascites is visible on ultrasound/CT in the large majority.
Clinical Features
- Ranges from asymptomatic to severe dyspnea and cough
- Rarely, rapid rupture into the pleural space can cause respiratory failure
- Thoracentesis: transudative in 94% (by Light's criteria; note Light's criteria can misclassify ~18% as exudates - use pleural/serum albumin ratio <0.6 as a more reliable marker in cirrhotics)
Management
- Sodium restriction + diuretics (mirrors ascites management)
- Repeated thoracentesis for symptom palliation (high recurrence risk)
- TIPS: reduces portal pressure, achieves response in 70-80% initially; limited by shunt occlusion, hepatic encephalopathy
- Chest tube drainage: NOT recommended - unacceptably high complication rate (protein/electrolyte loss, infection, renal failure)
- Video-assisted thoracoscopic surgery (VATS): diaphragmatic defect repair ± pleurodesis for refractory cases
- Indwelling pleural catheters (IPC): bridge to transplantation; empyema is the most common complication
- Liver transplantation: definitive - hepatic hydrothorax typically resolves; post-transplant outcomes are not inferior to those without hepatic hydrothorax
4. General Pulmonary Function Disturbances
(Murray & Nadel's Textbook of Respiratory Medicine)
These are background pulmonary abnormalities found broadly in advanced liver disease, even without HPS or POPH:
| Finding | Mechanism |
|---|---|
| Reduced DLCO | Most common PFT abnormality; reflects diffuse pulmonary microvascular changes |
| Restrictive physiology | Massive ascites elevating the diaphragm; improves with paracentesis |
| Hypocapnia / respiratory alkalosis | Hyperammonemia + hepatic encephalopathy driving hyperventilation; circulating estradiol and progesterone directly stimulating CNS respiratory centres; reflex hyperventilation from HPS/hydrothorax/POPH |
| Elevated (A-a)PO₂ | Found even in cirrhotic patients without overt HPS vs. matched controls |
| Obstructive physiology | Uncommon; when present, associated with smoking history rather than liver disease per se |
Summary Overview
| Complication | Key Identifier | Core Mechanism | Hallmark Sign | Definitive Treatment |
|---|---|---|---|---|
| HPS | Orthodeoxia + platypnea | Intrapulmonary vasodilation (NO, ET-1, VEGF) | Positive bubble echo (late left-sided microbubbles) | Liver transplantation |
| POPH | Exertional dyspnea + right heart failure | Pulmonary vasoconstriction + vascular remodeling | mPAP >20 on RHC + PVR >240 | PAH therapy ± liver Tx (if mPAP <45) |
| Hepatic Hydrothorax | Right-sided transudative effusion in cirrhosis | Diaphragmatic defects + ascites pressure gradient | Unilateral right pleural effusion, no cardiac disease | Liver transplantation; TIPS bridge |
| PFT disturbances | Reduced DLCO, respiratory alkalosis | Ascites, microvasculopathy, hyperammonemia | Hypocapnia on ABG | Treat underlying cause |
Acute on chronic liver
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
I now have comprehensive data from all four major textbooks. Here is the full structured response.
Acute-on-Chronic Liver Failure (ACLF)
ICD-10: K72.11
Sources: Robbins & Cotran Pathologic Basis of Disease | Robbins & Kumar Basic Pathology | Harrison's Principles of Internal Medicine 22e (2025) | Sleisenger & Fordtran's Gastrointestinal and Liver Disease
Definition
ACLF is a clinical syndrome characterised by acute decompensation of previously stable chronic liver disease (with or without cirrhosis), resulting in failure of one or more organ systems and high short-term mortality.
- Harrison's (2025): "Acutely decompensating cirrhosis with associated failure of one or more organ systems - liver, kidneys, brain, lung, circulatory system, and coagulation - analogous to sepsis syndrome."
- Sleisenger & Fordtran: "A condition in patients with underlying chronic liver disease with or without cirrhosis that is associated with mortality within 3 months in the absence of treatment of the underlying liver disease, liver support, or liver transplantation."
- Robbins: "Some individuals with stable but well-compensated, advanced chronic liver disease suddenly develop signs of acute liver failure...short-term mortality is around 50%."
Classification by Underlying Liver Disease (Sleisenger & Fordtran, Type A/B/C)
| Type | Underlying Disease |
|---|---|
| Type A | Chronic liver disease without cirrhosis |
| Type B | Underlying compensated cirrhosis |
| Type C | Underlying decompensated cirrhosis |
Epidemiology
-
5% of all hospitalizations in patients with cirrhosis are for ACLF, and incidence is rising
- Over two-thirds of hospitalized ACLF patients have an active infection
- In-hospital mortality ~50%
- ACLF mortality exceeds that of acute liver failure (ALF) after 1 week of hospitalization; unlike ALF, the high mortality risk in ACLF persists rather than returning to baseline at ~3 weeks
- ~50% of ACLF patients listed for liver transplantation are delisted or dead within 6 months
Pathophysiology
Core Mechanism: Systemic Inflammation + Immune Dysregulation
(Sleisenger & Fordtran)
The gut microbiome is central. An acute precipitant (e.g., alcohol binge, infection) triggers:
- Gut bacterial translocation → pathogen-associated molecular patterns (PAMPs) enter the circulation
- Marked systemic inflammatory response - more pronounced than in simple decompensated cirrhosis; levels of markers of cell death are also markedly elevated
- Sterile inflammation from hepatocyte death (e.g., alcohol-induced) superimposed on infection-driven inflammation
- Innate immune suppression: patients develop significant suppression of the innate immune system, representing "failed immune tolerance"
- Compensatory anti-inflammatory response (CARS): immunosuppression with enhanced susceptibility to secondary infections → organ failure cascade
Geographic variation:
- East: HBV reactivation, HEV superinfection in chronic liver disease, alcoholic hepatitis dominate
- West: Bacterial infection and alcoholic hepatitis dominate
Precipitating Factors (Harrison's 2025)
| Category | Specific Triggers |
|---|---|
| Direct hepatic insults | Alcoholic hepatitis (most common in West), new/flaring viral hepatitis (HBV, HCV, HEV, HAV), autoimmune hepatitis flare, drug-induced liver injury (DILI) |
| Systemic/extrahepatic | Bacterial or fungal infection (most common overall), GI bleeding, postoperative state |
| Special triggers (Robbins) | HDV superinfection in chronic HBV; emergence of drug-resistant viral mutants in suppressed HBV; ascending cholangitis in PSC or fibrocystic liver disease; MASH decompensation from rapid weight loss/malnutrition; sepsis with attendant hypotension; acute cardiac failure; superimposed drug/toxic injury; occult malignancy (HCC, cholangiocarcinoma, metastases) |
Clinical Features
Patients present with features of systemic inflammatory response syndrome (SIRS):
- Fever, tachycardia, tachypnea, leukocytosis
Combined with manifestations of organ failure:
Organ Failure Manifestations in ACLF (Sleisenger & Fordtran, Table 74.2)
| Organ | Manifestation |
|---|---|
| Liver | Loss of metabolic function: hypoglycaemia, lactic acidosis, hyperammonaemia, coagulopathy |
| Kidneys | Type 1 hepatorenal syndrome (HRS-1) or need for renal replacement therapy |
| Brain | Hepatic encephalopathy grade 3-4 |
| Circulation | Need for vasopressor support |
| Lungs | Acute lung injury (ALI) / ARDS requiring ventilatory support |
| Adrenal gland | Hypotension (adrenal insufficiency) |
| Bone marrow | Suppression (pancytopenia) |
Prognosis - Organ Failure Scoring (Sleisenger & Fordtran)
The number of organ failures is the primary determinant of prognosis:
| Organ failures | In-hospital mortality |
|---|---|
| 2 | 27% |
| 3 | 65% |
| 4 | 97% |
The presence of 2 or more extrahepatic organ failures is specifically associated with poor prognosis.
Scoring systems used:
- CLIF-C ACLF score (European/CANONIC study) - based on organ failure grading
- SOFA (Sequential Organ Failure Assessment) - more accurately reflects ICU prognosis than Child-Pugh or MELD scores in the ICU setting (Sleisenger & Fordtran)
- NACSELD score (North American)
Management (Harrison's 2025 + Sleisenger & Fordtran)
General Principles
- Managed by a multidisciplinary team with critical care and liver transplantation expertise
- Search for and treat all precipitating causes
- Determine if ICU care is needed
- Immediate referral for liver transplantation evaluation
Specific Interventions (Table 74.3, Sleisenger & Fordtran)
| Pathophysiology Target | Intervention |
|---|---|
| Liver failure | Hepatic regenerative therapy; artificial/bioartificial liver support as bridge to LT |
| Alcoholic hepatitis | Glucocorticoids (prednisolone) |
| HBV flare | Antiviral agents (entecavir, tenofovir) |
| Infection | Targeted antibiotics |
| Extrahepatic organ failure | Organ-specific support (vasopressors, RRT, ventilation) |
Key Notes on Specific Treatments
- Hepatorenal syndrome (HRS-1): poor response to terlipressin in advanced ACLF
- Liver support devices (MARS, Prometheus): bioartificial liver support has not been proven effective in ACLF-specific trials; no demonstrated mortality benefit
- Granulocyte-colony stimulating factor (G-CSF) + erythropoietin: combination shown to decrease mortality risk in decompensated cirrhosis; hepatic regenerative therapy is a promising direction
- Routine prophylactic antibiotics in all cirrhotics are not currently recommended
Transition to Palliative Care (Harrison's 2025)
- If after 3-7 days of ICU support, there are 4 or more organ failures and liver transplantation is not an option, transition to palliative care should be considered
Liver Transplantation in ACLF
- LT offers the only hope of long-term survival (Sleisenger & Fordtran)
- Patients with multiple organ failures may be too sick for transplantation
- In selected patients with alcohol-associated hepatitis who undergo LT, long-term results have been good
- Na-MELD score ≥15 is used to identify patients likely to benefit from transplantation
- ACLF patients may receive special MELD exception points for expedited listing
Summary
Stable chronic liver disease (cirrhosis)
↓ Precipitant (infection, alcohol, viral, drugs)
Acute decompensation
↓ Systemic inflammation + immune dysregulation
Multiorgan failure
↓
ACLF (28-day mortality 20-70% depending on organ failure count)
↓
Treatment: Remove precipitant + organ support + LT (if eligible)
Sources: Robbins & Cotran Pathologic Basis of Disease, p. 780 | Robbins & Kumar Basic Pathology | Harrison's Principles of Internal Medicine 22e (2025) | Sleisenger & Fordtran's Gastrointestinal and Liver Disease, Ch. 74
Drug and toxin induced liver injury
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
I now have comprehensive data from Robbins (both editions), Sleisenger & Fordtran, and supplementary sources. Here is the full structured answer.
Drug- and Toxin-Induced Liver Injury (DILI / TILI)
ICD-10: K71 (Drug-induced liver disease)
Sources: Robbins & Cotran Pathologic Basis of Disease | Robbins & Kumar Basic Pathology | Sleisenger & Fordtran's Gastrointestinal and Liver Disease (Ch. 88) | Lippincott's Pharmacology
Overview
The liver is the principal drug-metabolising and detoxifying organ in the body. Its position in the portal circulation means it is the first organ exposed to ingested drugs, toxins, and gut-derived microbial products. Drug- and toxin-induced liver injury is a major cause of acute liver failure in the United States.
Key epidemiological facts (Robbins; Sleisenger & Fordtran):
- Drugs account for >50% of cases of ALF referred to specialist units in the USA
- 10% of patients with DILI die or require liver transplantation
- 17% develop chronic liver disease
- Drugs account for 43% of severe hepatitis in patients aged >50 years
- Herbal and dietary supplements account for >25% of DILI in some countries (e.g., South Korea)
Hepatic Drug Metabolism - Why the Liver is Vulnerable
(Sleisenger & Fordtran)
The liver eliminates drugs through three coordinated phases:
| Phase | Process | Key Players |
|---|---|---|
| Phase 1 | Oxidation, reduction, hydrolysis - converts lipophilic drugs to reactive intermediates | Cytochrome P-450 (CYP) enzymes - >20 isoforms in human liver |
| Phase 2 | Conjugation with glucuronide, sulfate, glutathione - renders metabolites water-soluble | UGTs, sulfotransferases, glutathione-S-transferases |
| Phase 3 | Energy-dependent biliary or renal excretion of metabolites | Membrane transporters (MDR, MRP, BSEP) |
The critical CYP2E1 pathway and NAPQI:
- CYP2E1 (localised in acinar zone 3 - centrilobular hepatocytes) catalyses oxidation of acetaminophen to NAPQI (N-acetyl-p-benzoquinone imine) - a highly reactive electrophilic and oxidising metabolite
- Under normal doses, NAPQI is rapidly conjugated and detoxified by glutathione
- At high doses or with glutathione depletion, free NAPQI covalently binds hepatocyte proteins and mitochondrial enzymes → zone 3 (centrilobular) necrosis
- CYP2E1 is concentrated in zone 3, explaining why acetaminophen toxicity is always centrilobular
CYP inducers that amplify drug toxicity:
Rifampicin, phenytoin, isoniazid, tobacco smoke, and ethanol all induce the CYP system and can markedly exacerbate the toxicity of other drugs by increasing generation of toxic metabolites.
Classification of DILI: Predictable vs. Idiosyncratic
(Robbins & Kumar Basic Pathology; Robbins & Cotran)
| Feature | Intrinsic / Predictable (Direct) | Idiosyncratic / Unpredictable |
|---|---|---|
| Dose-dependence | Yes - affects all individuals in a dose-dependent fashion | No - occurs in susceptible individuals regardless of dose |
| Frequency | Affects everyone exposed above a threshold dose | Rare (1 in 1,000 to 1 in 100,000 exposed) |
| Onset | Predictable; often rapid | Variable - 1 to 3 months of exposure (up to years) |
| Mechanism | Direct chemical toxicity of drug or reactive metabolite | Hypersensitivity (immune-mediated) OR metabolic idiosyncrasy |
| Prototype | Acetaminophen, CCl₄, Amanita phalloides | Isoniazid, halothane, chlorpromazine, nitrofurantoin |
Mechanisms of Hepatocellular Injury
(Sleisenger & Fordtran)
1. Direct / Intrinsic Toxicity
- Drug or its reactive metabolite directly injures the hepatocyte
- Mitochondrial injury is a primary target: NAPQI (acetaminophen) directly damages mitochondrial enzymes; mitochondrial permeability transition releases Ca²⁺, activates JNK and GSK-3β signalling → further mitochondrial dysfunction
- Necrosis pathway: Drug injury de-energises mitochondria → ATP depletion → cell swelling → membrane rupture → necrosis (when ATP is absent) or apoptosis (when ATP is preserved)
- Reactive oxygen species (ROS) generate oxidative stress and trigger cell death
2. Immune-Mediated (Hypersensitivity) Idiosyncrasy
- Drug or its metabolite acts as a hapten, covalently binding to cellular proteins → creates new immunogen
- Triggers CD8+ cytotoxic T-cell response and antibody formation against hepatocytes
- Features: fever, rash, eosinophilia, arthralgia (DRESS syndrome), positive rechallenge
- Prototype: halothane hepatitis (fatal immune hepatitis after repeated exposure)
3. Metabolic Idiosyncrasy
- Drug is converted by an unusual metabolic pathway in a genetically susceptible individual to a toxic metabolite
- No overt immune features
- Prototype: isoniazid - slow acetylators (NAT2 variant) accumulate toxic acetylhydrazine metabolites → hepatitis
4. Mitochondrial Toxicity
- Direct impairment of mitochondrial oxidative phosphorylation → microvesicular steatosis (fatty change with small cytoplasmic fat droplets = Reye-like picture)
- Drugs: valproic acid, tetracycline, zidovudine, didanosine, zalcitabine, fialuridine
- Results from impaired β-oxidation of fatty acids with fat accumulation in small vacuoles
Patterns of Morphological Injury
(Robbins & Cotran, Table 18.5 - full classification)
| Pattern | Morphological Findings | Causative Agents |
|---|---|---|
| Hepatocellular necrosis - zone 3 (centrilobular) | Confluent necrosis with sparse inflammation | Acetaminophen, halothane, CCl₄ |
| Acute hepatitis - inflammation dominant | Lymphocytic ± plasma cell ± eosinophil infiltrate; spotty/confluent necrosis | Isoniazid, antimicrobials, anticonvulsants, methyldopa, phenytoin, PD-1/PD-L1/CTLA-4 inhibitors (immune checkpoint inhibitors) |
| Cholestatic (bland) | Hepatocellular cholestasis without inflammation | Contraceptive and anabolic steroids, antibiotics, antiretrovirals (ART) |
| Cholestatic hepatitis | Cholestasis + lobular necroinflammatory activity ± bile duct destruction | Antibiotics, phenothiazines (chlorpromazine), statins |
| Chronic hepatitis | Portal lymphocytic/lymphoplasmacytic inflammation ± fibrosis | Nitrofurantoin, NSAIDs, methyldopa |
| Macrovesicular steatosis | Large fat droplets in hepatocytes | Ethanol, corticosteroids, methotrexate, TPN |
| Microvesicular steatosis | Diffuse small fat droplets (Reye-like) | Valproate, tetracycline, aspirin (Reye syndrome), zidovudine, ART |
| Steatohepatitis | Fat + ballooning + Mallory-Denk hyaline | Ethanol, amiodarone, irinotecan, tamoxifen |
| Fibrosis / cirrhosis | Periportal and pericellular fibrosis | Alcohol, methotrexate, enalapril, vitamin A/retinoids |
| Granulomas - non-caseating epithelioid | Epithelioid cell granulomas without caseous necrosis | Sulfonamides, amiodarone, isoniazid |
| Fibrin ring granulomas | Fibrin ring granulomas (like Q fever) | Allopurinol |
| Sinusoidal obstruction syndrome (SOS/VOD) | Obliteration of central veins (formerly veno-occlusive disease) | High-dose chemotherapy, pyrrolizidine alkaloids (bush teas) |
| Budd-Chiari syndrome | Hepatic vein thrombosis → outflow obstruction | Oral contraceptives |
| Peliosis hepatitis | Blood-filled non-endothelium-lined cavities in parenchyma | Anabolic steroids, tamoxifen |
| Hepatocellular adenoma | Benign hepatocellular neoplasm | Oral contraceptives, anabolic steroids |
| Hepatocellular carcinoma | Malignant hepatocellular tumour | Alcohol, Thorotrast |
| Angiosarcoma | Malignant endothelial tumour | Thorotrast, vinyl chloride, arsenic |
Prototype Drug: Acetaminophen (Paracetamol) - Direct Hepatotoxin
Most common cause of ALF requiring transplantation in the USA (Robbins)
Mechanism
Acetaminophen
↓ (CYP2E1, CYP3A4 - concentrated in zone 3)
NAPQI (N-acetyl-p-benzoquinone imine)
↓ (normal dose) ↓ (overdose / glutathione depleted)
Conjugated with → Free NAPQI
glutathione (safe) ↓
Covalently binds hepatocyte proteins
+ Mitochondrial enzyme damage
+ JNK / GSK-3β activation
↓
Zone 3 (centrilobular) NECROSIS
Factors that increase toxicity:
- Fasting (depletes glutathione)
- Chronic alcohol use (induces CYP2E1 + depletes glutathione) - alcoholics develop toxicity at therapeutic doses
- Enzyme inducers (rifampicin, phenytoin, isoniazid)
Antidote: N-acetylcysteine (NAC) - replenishes glutathione; also has direct anti-inflammatory and antioxidant effects. Effective if given within 8-10 hours; can still be beneficial up to 24 hours post-ingestion.
Prototype Drug: Isoniazid (INH) - Metabolic Idiosyncratic Hepatotoxin
- CYP and NAT2 metabolise INH → acetylhydrazine and other reactive metabolites
- Slow acetylators (NAT2 variant): accumulate toxic acetylhydrazine → hepatotoxicity
- Fast acetylators: may generate more hydrazine (alternative toxic metabolite)
- Hepatitis develops in ~1% of patients; rarely fulminant (preventable deaths still occur)
- Enzyme inducers (rifampicin, phenytoin, alcohol) increase risk
- Concomitant rifampicin accelerates INH metabolism to hydrazine
Prototype Drug: Halothane - Immune-Mediated Hepatotoxin
- Minor halothane metabolism by CYP2E1 generates trifluoroacetyl chloride (TFA) → covalently binds liver proteins → neoantigens
- Prior sensitisation required: fatal hepatitis classically occurs on repeated exposure
- Characterised by fever, eosinophilia, elevated LFTs appearing 1-2 weeks post-anaesthesia
- Mostly replaced by sevoflurane and desflurane (much lower metabolic rate)
Risk Factors for DILI (Sleisenger & Fordtran, Table 88.2)
| Factor | Effect |
|---|---|
| Drug dose ≥50 mg/day | Increases risk for both intrinsic and some idiosyncratic reactions |
| Age >50 years | Increased risk (reduced drug metabolism, more polypharmacy) |
| Female sex | Higher risk for some reactions (e.g., halothane, nitrofurantoin, autoimmune-type) |
| Genetic polymorphisms | NAT2 (isoniazid), CYP2D6, HLA alleles |
| Alcohol use | CYP2E1 induction; glutathione depletion |
| Chronic HCV | Increased risk with several drug groups |
| Chronic HBV | Risk with antituberculous drugs; HBV reactivation risk with immunosuppressives |
| Malnutrition/fasting | Depletes glutathione; worsens acetaminophen toxicity |
| Pregnancy | Increased risk of tetracycline and valproate hepatotoxicity |
| Prior drug reactions | Increased susceptibility to related compounds |
Diagnosis
(Sleisenger & Fordtran)
DILI is a diagnosis of exclusion. The key components are:
- Temporal association: Drug exposure precedes onset; liver injury resolves (usually) on drug withdrawal
- Exclusion of other causes: Viral hepatitis (including HEV), autoimmune hepatitis, biliary obstruction, vascular disorders
- Hy's Rule (FDA guideline): ALT ≥3× ULN + bilirubin ≥2× ULN (without ALP elevation >2× ULN) predicts ~10% risk of ALF in the exposed population - signals a drug's potential to cause serious hepatotoxicity
- R ratio = (ALT/ULN) ÷ (ALP/ULN): R >5 = hepatocellular pattern; R <2 = cholestatic; R 2-5 = mixed
- RUCAM (Roussel-Uclaf Causality Assessment Method) / RECAM: Standardised causality scoring tool
- Rechallenge: Positive if ALT or ALP rises ≥2-fold on re-exposure (deliberate rechallenge rarely justified)
- Extrahepatic features: Rash, fever, eosinophilia, lymphadenopathy support immune mechanism (DRESS syndrome)
- Liver biopsy: When diagnosis is uncertain or drug-induced chronic hepatitis/cholestasis is suspected
- LiverTox database (livertox.nih.gov): NIH resource for drug-specific hepatotoxicity profiles
Management
| Situation | Action |
|---|---|
| All DILI | Discontinue the offending drug immediately |
| Acetaminophen toxicity | N-acetylcysteine (NAC) - within 8-24 hours of ingestion |
| Severe hepatitis / ALF | Supportive ICU care; urgent liver transplant evaluation |
| Cholestatic DILI | Drug withdrawal; ursodeoxycholic acid may help (limited evidence) |
| Immune-type DILI | Drug withdrawal; corticosteroids in severe cases (immune checkpoint inhibitor-related) |
| Prevention | Drug dose monitoring; avoid enzyme inducers with known hepatotoxins; patient education; hepatitis vaccinations |
Summary of Key DILI Examples by Pattern
| Drug | Pattern | Mechanism | Key Feature |
|---|---|---|---|
| Acetaminophen | Zone 3 necrosis | Direct (NAPQI/CYP2E1) | Dose-dependent; glutathione depletion; NAC antidote |
| Isoniazid | Acute/chronic hepatitis | Metabolic idiosyncrasy (acetylhydrazine) | Slow NAT2 acetylators; fulminant risk |
| Halothane | Massive necrosis | Immune (TFA neoantigens) | Repeated exposure; CYP2E1-mediated sensitisation |
| Chlorpromazine | Bland cholestasis | Metabolic idiosyncrasy | Slow metabolizers; jaundice ± eosinophilia |
| Amiodarone | Steatohepatitis + Mallory hyaline | Mitochondrial toxicity + phospholipidosis | Resembles alcoholic hepatitis; granulomas possible |
| Methotrexate | Fibrosis / cirrhosis | Direct (dose-dependent) | Periportal/pericellular fibrosis; monitor with liver biopsy |
| Valproate | Microvesicular steatosis | Mitochondrial inhibition | Reye-like picture; children at risk |
| Oral contraceptives | Bland cholestasis / Budd-Chiari / hepatocellular adenoma | Oestrogen effect on bile transport + venous thrombosis | Cholestasis, venous thrombosis, benign liver tumours |
| Anabolic steroids | Cholestasis / peliosis / adenoma / HCC | Androgen receptor mediated | Blood-filled peliotic cavities; HCC risk |
| Amanita phalloides | Massive necrosis | Direct (amatoxin inhibits RNA polymerase II) | Mushroom poisoning; no antidote |
| Nitrofurantoin | Chronic hepatitis / fibrosis | Immune | May resemble autoimmune hepatitis |
| Immune checkpoint inhibitors (anti-PD-1, anti-CTLA-4) | Acute hepatitis | Immune activation | Increasingly recognised; treat with corticosteroids |
Sources: Robbins & Cotran Pathologic Basis of Disease, Ch. 18, Table 18.5, p. 781-782 | Robbins & Kumar Basic Pathology, Table 14.3 | Sleisenger & Fordtran's GI and Liver Disease, Ch. 88
This is a shared conversation. Sign in to Orris to start your own chat.