Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathogenesis
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have comprehensive, authoritative content from Murray & Nadel's Textbook of Respiratory Medicine. Here is the full, detailed explanation:
Mechanism of Acute Respiratory Distress Syndrome (ARDS)
ARDS is a syndrome of acute, non-cardiogenic pulmonary edema caused by increased alveolar-capillary permeability, resulting in protein-rich fluid flooding the airspaces, profound hypoxemia, and bilateral radiographic infiltrates. It carries a mortality commonly exceeding 30%.
1. Triggers and Classification of Injury
ARDS arises from two broad categories of insult:
| Direct (Pulmonary) Injury | Indirect (Nonpulmonary) Injury |
|---|---|
| Pneumonia (bacterial, viral - COVID-19, influenza) | Sepsis (most common cause overall) |
| Aspiration of gastric contents | Major trauma |
| Pulmonary contusion | Multiple blood transfusions |
| Toxic inhalation | Pancreatitis |
| Near-drowning | Cardiopulmonary bypass |
| Reperfusion injury (e.g., post-lung transplant) | Drug overdose |
- Murray & Nadel's Textbook of Respiratory Medicine, Table 134.2
2. Core Pathophysiology: Increased Alveolar-Capillary Permeability
Unlike cardiogenic pulmonary edema (driven by elevated hydrostatic pressure), ARDS is defined by a permeability defect in the alveolar-capillary barrier. Both the microvascular endothelium and the alveolar epithelium are disrupted.
Key consequences:
- Protein-rich, exudative fluid floods the alveolar spaces
- Alveolar fluid clearance is impaired (loss of epithelial pumping function)
- Respiratory system compliance falls dramatically
- Right-to-left shunting develops, causing profound hypoxemia
- Dead space ventilation increases, driving up minute ventilation
- Pulmonary hypertension develops via hypoxic vasoconstriction, intravascular fibrin deposition, and compression from positive-pressure ventilation
Multiple mechanisms drive epithelial and endothelial cell death: necrosis, apoptosis, activation of coagulation cascades, and mechanical stretch injury. Loss of endothelial barrier integrity is both necessary and sufficient for ARDS to develop.
3. Pathological Stages (Diffuse Alveolar Damage - DAD)
The histological hallmark of ARDS is diffuse alveolar damage (DAD), which progresses through three overlapping phases:
Phase 1 - Exudative (Days 1-7)
- Hyaline membranes form in alveoli (composed of cellular debris, proteins, surfactant components)
- Protein-rich fluid fills airspaces
- Widespread epithelial disruption
- Massive neutrophil infiltration of interstitium and airspaces
- Elevated N-terminal procollagen peptide III in BAL fluid detectable within 24 hours (early signal of fibrogenesis)
Phase 2 - Proliferative (Days 7-21)
- Hyaline membranes are reorganized
- Fibrosis begins to appear
- Obliteration of pulmonary capillaries
- Deposition of interstitial and alveolar collagen
- Decrease in neutrophil number and extent of edema
Phase 3 - Fibrotic (>2 weeks, subset of patients)
- Frank pulmonary fibrosis in patients with persistent ARDS
The CT images below illustrate these phases sequentially:
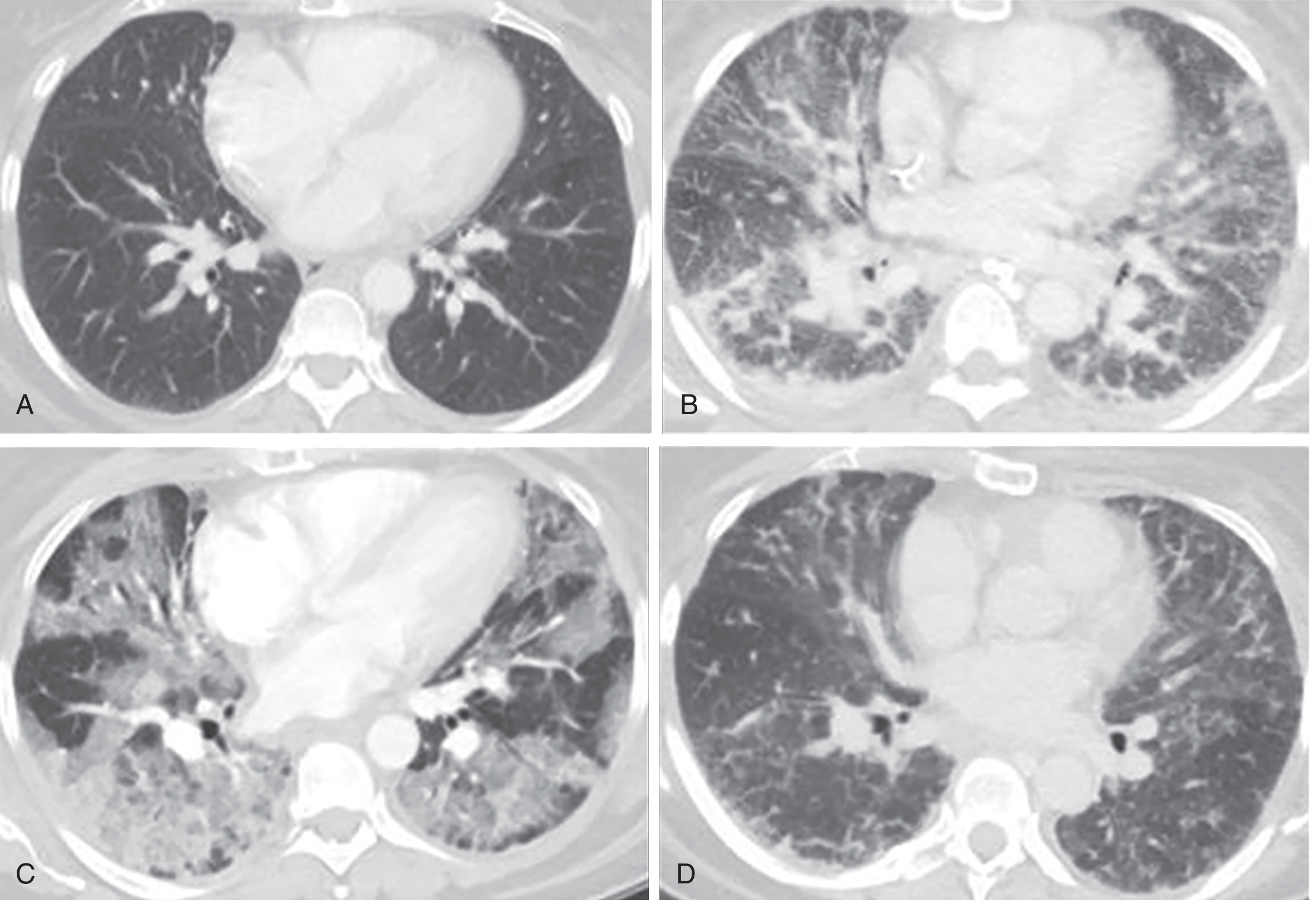
4. The Central Role of Neutrophils
Neutrophil accumulation in the lung microvasculature is one of the earliest and most consistent hallmarks of ARDS.
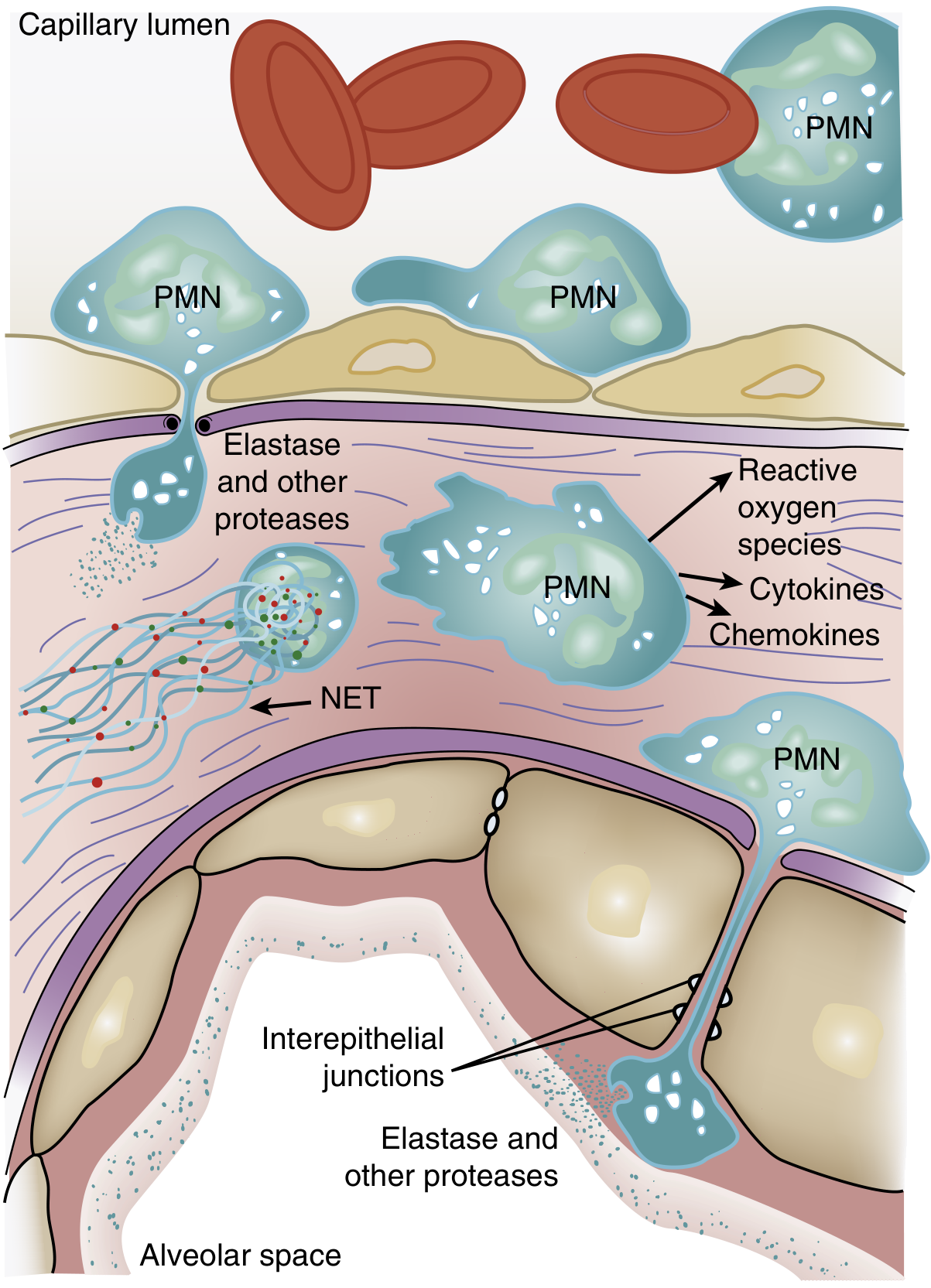
Mechanism of Neutrophil-Mediated Injury
Step 1 - Sequestration:
The average pulmonary capillary is narrower than the average neutrophil diameter. Activated neutrophils become "stiff" (via actin cytoskeleton remodeling), cannot deform to pass through, and get sequestered. This causes the characteristic transient leukopenia that precedes hypoxemia. Once sequestered, neutrophils can transmigrate without the usual requirement for L-selectin and beta-2 integrins.
Step 2 - Activation:
Triggers include lipopolysaccharide (LPS), cytokines, and neutrophil-platelet interactions. Key intracellular signaling involves p38 mitogen-activated protein kinase (MAPK), which is activated by LPS and drives TNF-alpha production and macrophage inflammatory protein-2 release.
Step 3 - Tissue damage via four weapons:
- Reactive oxygen species (ROS) - oxidative damage to epithelial and endothelial cells
- Proteolytic enzymes - especially neutrophil elastase (NE) and matrix metalloproteinases (MMPs). NE degrades surfactant protein A, ECM components, and disrupts interepithelial tight junctions
- Cytokines/chemokines - TNF-alpha, IL-1beta amplify the inflammatory cascade and recruit more neutrophils
- Neutrophil Extracellular Traps (NETs) - web-like structures of DNA, histones, myeloperoxidase, NE, and cathepsin G. In sepsis, large-scale NET release causes endothelial damage and thrombosis. Animal models show that DNase treatment (which degrades NETs) attenuates lung injury and lowers IL-6/TNF levels. BAL fluid from ARDS patients shows NET concentration inversely correlated with degree of ARDS.
Note: ARDS can occur in profoundly neutropenic patients, suggesting alveolar macrophages may serve as an alternative driver of injury in some cases.
5. Surfactant Dysfunction
In healthy lungs, surfactant (predominantly dipalmitoylphosphatidylcholine and phosphatidylglycerol) reduces alveolar surface tension and prevents collapse. In ARDS:
- The proportion of large (active) to small (inactive) surfactant aggregates is diminished
- Surfactant production by type II pneumocytes falls
- Plasma proteins that leak into alveoli interfere with surfactant function
- Neutrophil elastase directly degrades surfactant protein A (confirmed in BAL fluid from ARDS patients)
- Phospholipase A2 (relevant in pancreatitis-associated ARDS) enzymatically degrades surfactant and promotes alveolar collapse and increased vascular permeability
The result is alveolar collapse and atelectasis, worsening hypoxemia and reducing compliance further.
6. Inflammation and Coagulation Crosstalk
Inflammation and coagulation are tightly linked in ARDS:
- TNF-alpha increases thrombin and fibrin formation, stimulates tissue factor expression on endothelial cells, and inhibits fibrinolysis
- Fibrin fragments are chemotactic for neutrophils - a self-amplifying loop
- Intravascular fibrin deposition contributes to pulmonary hypertension
- Activated protein C (endogenous anticoagulant) has anti-inflammatory effects including IL-6 downregulation and neutrophil activation attenuation
- Other key mediators include IL-8 (potent neutrophil chemoattractant), platelet-activating factor, adhesion molecules, and reactive oxygen species
7. Cytokine-Driven "Cytokine Storm" Amplification
In severe ARDS (particularly sepsis-associated), a self-amplifying cytokine cascade drives progression:
- TNF-alpha and IL-1beta are produced by macrophages and activated neutrophils early in the response
- These stimulate further neutrophil recruitment, endothelial activation, and coagulation
- IL-8 acts as the principal chemokine drawing more neutrophils to the lung
- In pancreatitis-associated ARDS specifically: TNF-alpha and IL-8 augment neutrophil recruitment, while phospholipase A2 and elastase from pancreatic tissue directly damage the lung; reactive oxygen species and platelet-activating factor contribute further
8. Functional Consequences
| Mechanism | Physiological Effect |
|---|---|
| Alveolar flooding with exudate | Reduced FRC, decreased compliance |
| Alveolar collapse/atelectasis | Right-to-left shunt, refractory hypoxemia |
| Interstitial edema | Increased dead space ventilation |
| Vascular occlusion (fibrin, hypoxic vasoconstriction) | Pulmonary hypertension |
| Surfactant inactivation | Further atelectasis, increased work of breathing |
| Ventilator-induced lung injury (VILI) | Biotrauma - barotrauma/volutrauma amplifies inflammatory mediator release |
Summary Diagram of Mechanism
Precipitating Insult (Sepsis / Pneumonia / Aspiration / Trauma)
↓
Systemic / Local Inflammatory Activation
↓
Cytokines (TNF-α, IL-1β, IL-8) + Complement + Coagulation activation
↓
Neutrophil sequestration in pulmonary capillaries
↓
Neutrophil transmigration → ROS + Elastase + NETs + Cytokines
↓
Alveolar-capillary barrier disruption (endothelium + epithelium)
↓
Protein-rich exudate floods alveoli + Surfactant inactivation
↓
Diffuse Alveolar Damage (DAD) = Hyaline membranes + Atelectasis
↓
Hypoxemia (shunt) + Decreased compliance + Pulmonary hypertension
↓
Exudative → Proliferative → Fibrotic phases
Sources:
- Murray & Nadel's Textbook of Respiratory Medicine, Chapter 134 (ETIOLOGY AND PATHOGENESIS OF ARDS, THE ALVEOLAR-CAPILLARY MEMBRANE, NEUTROPHILS AND OTHER INFLAMMATORY MEDIATORS, INFLAMMATION AND COAGULATION)
- Goldman-Cecil Medicine, Chapter 91 (Mechanical Ventilation in ARDS)
This is a shared conversation. Sign in to Orris to start your own chat.