Explain retinitis pigmentosa in detail
Reading File
Finding Sources
Searching PubMed
"retinitis pigmentosa"[MeSH Terms]
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have comprehensive information from multiple authoritative textbooks. Let me compose a thorough, well-organized answer.
Retinitis Pigmentosa - Detailed Overview
Definition
Retinitis pigmentosa (RP) is a general term for a clinically and genetically diverse group of inherited, diffuse retinal degenerative diseases that initially and predominantly affect rod photoreceptors, with subsequent degeneration of cones and the retinal pigment epithelium (RPE). Despite its name, RP is not an inflammatory condition - the term "retinitis" is a misnomer from an era when these disorders were incorrectly presumed to be inflammatory.
Epidemiology
- Most common hereditary retinal degeneration worldwide
- Prevalence: approximately 1 in 3,000 to 1 in 5,000 individuals
- Bilateral and roughly symmetrical in presentation
- Age of onset, rate of progression, and extent of visual loss are often related to the inheritance pattern
Fundoscopic Appearance
The classic hallmark image of RP - showing the characteristic bone-spicule pigmentation distributed around the peripheral and mid-peripheral retina, with attenuated vessels and a pale optic disc:
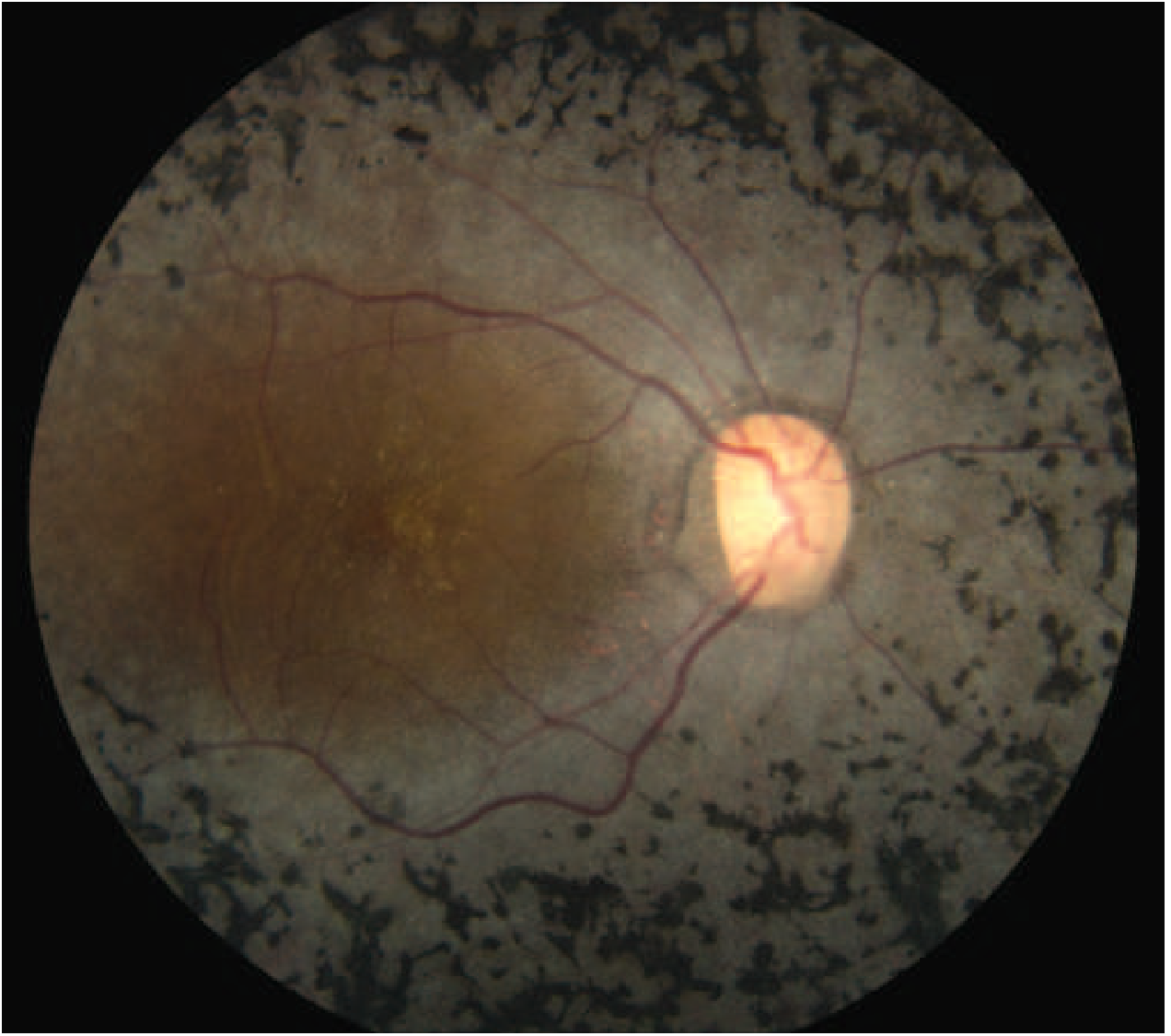
Classic fundus appearance of RP with bone-spicule pigmentation. Note the sparse, attenuated retinal vessels and pale optic disc, with relative macular sparing early in disease - Harrison's Principles of Internal Medicine 22E
Genetics and Molecular Basis
RP can be sporadic or inherited in several patterns:
| Inheritance Pattern | Frequency | Key Features |
|---|---|---|
| Autosomal recessive (AR) | Most common | Severe, early onset; night blindness presents early in life |
| Autosomal dominant (AD) | Intermediate | Milder, more gradual onset in adult life; variable penetrance; less severe visual loss |
| X-linked recessive | Rarest but most severe | Early onset similar to AR; female carriers show "salt-and-pepper" fundus |
| Sporadic | Variable | No other known affected family members; may still be AD/AR/X-linked |
Key genes involved:
- RHO (rhodopsin): Most cases of AD-RP; encodes the rod photopigment central to phototransduction
- RPGR (retinitis pigmentosa GTPase regulator): ~90% of X-linked RP; encodes a protein localised in the connecting cilium of rod photoreceptors
- Peripherin: A glycoprotein in photoreceptor outer segments
- USH2A, EYS: AR forms
- Over 100 gene loci are implicated in non-syndromic RP; despite this, approximately half of affected individuals have no identified molecular genetic abnormality
Pathogenic mechanisms fall into four categories:
- Defects in the phototransduction cascade (e.g., rhodopsin mutations)
- Defects in the retinoid cycle
- Defects in photoreceptor structure
- Other biological functions of the photoreceptor and RPE
Pathophysiology
- Rod photoreceptors die via apoptosis as the primary pathological event
- Cone cell death is partly mediated through activation of RIP kinase
- Loss of rods leads to progressive night blindness and peripheral visual field loss
- As cones are subsequently lost, central visual acuity deteriorates
- Retinal atrophy leads to:
- Constriction of retinal arterioles
- Optic nerve head atrophy ("waxy pallor" of the optic disc)
- Accumulation of retinal pigment around blood vessels - producing the "bone-spicule" pattern that gives the disease its name
Clinical Features
Symptoms (in order of typical appearance)
- Nyctalopia (night blindness) - usually the first and most prominent symptom; also dark adaptation difficulties
- Peripheral visual field loss - gradual constriction; patients may bump into objects
- Ring scotoma - characteristic mid-peripheral ring of visual loss
- Photopsia - not uncommon (flashes of light)
- Reduced central vision - typically a later feature, unless a cataract develops early
Signs
The Classic Triad (Diagnostic):
- Bone-spicule retinal pigmentation - bilateral mid-peripheral intraretinal perivascular "bone-spicule" pigmentary changes and RPE atrophy; may be absent early
- Arteriolar attenuation - narrowing of retinal blood vessels
- Waxy (pale) optic disc - optic nerve pallor with a distinctive waxy quality
Additional signs:
- Visual acuity may be normal initially (contrast sensitivity affected earlier)
- Progressive visual field loss, typically a ring scotoma that progresses to a small central tunnel
- Vitreous cells (most consistent sign per Wills Eye Manual)
- Areas of RPE depigmentation or atrophy
- Posterior subcapsular cataract (common association)
- Cystoid macular edema (CME)
- Epiretinal membrane (ERM)
Investigations
- Electroretinogram (ERG): Usually markedly reduced or extinguished - shows no electrical evidence of retinal function in advanced disease; the most important functional test
- Visual fields (perimetry): Ring scotoma progressing to tunnel vision
- Dark adaptometry: Impaired dark adaptation
- Optical coherence tomography (OCT): Detects retinal thinning, CME, or ERM
- Genetic testing: Identifies specific mutations (RHO, RPGR, etc.); guides prognosis and gene therapy eligibility
- Family pedigree: Should be prepared; important for genetic counselling
Syndromic RP (20-30% of cases)
RP is associated with systemic disorders in approximately 20-30% of cases. These are usually AR or mitochondrial in inheritance:
| Syndrome | Key Systemic Features |
|---|---|
| Usher syndrome (AR; accounts for ~5% of profound childhood deafness) | Sensorineural deafness + vestibular dysfunction. Type I: most severe; Type III: progressive hearing loss with late-onset RP |
| Bardet-Biedl syndrome (AR) | Obesity, polydactyly, renal anomalies, hypogonadism, cognitive impairment |
| Refsum disease (AR) | Phytanic acid accumulation; ichthyosis, peripheral neuropathy, cerebellar ataxia, anosmia |
| Bassen-Kornzweig (abetalipoproteinemia) (AR) | Fat malabsorption, acanthocytosis, spinocerebellar ataxia; low vitamins A/D/E/K |
| Kearns-Sayre syndrome (mitochondrial) | Ophthalmoplegia, cardiac conduction defects, ataxia |
| Leber congenital amaurosis | Severe early-onset rod-cone dystrophy; ~5% of all RP; presents at birth/infancy |
| NARP syndrome (mitochondrial) | Neuropathy, ataxia, retinitis pigmentosa |
| Olivopontocerebellar degeneration | Cerebellar/brainstem atrophy |
Drug-Induced Mimics
Certain drugs can cause a toxic retinopathy resembling RP:
- Chloroquine / Hydroxychloroquine - produces a characteristic "bull's-eye maculopathy"; patients on long-term hydroxychloroquine need regular OCT screening
- Thioridazine (and other phenothiazines)
Management
There is currently no cure for RP. Management is aimed at slowing progression and optimising remaining vision:
Pharmacological
- Vitamin A palmitate (15,000 IU/day): Some evidence supports slowing of midperipheral visual field loss in nonsmokers; if initiated, monitor liver function tests and vitamin A levels (Goldman-Cecil, Harrison's, Wills Eye)
- Lutein (12 mg/day): Combined with vitamin A, may have some benefit; literature remains inconclusive
- Low phytanic acid diet (in Refsum disease) and vitamin supplementation + low-fat diet (in Bassen-Kornzweig) can retard progression
Optical and Low Vision Aids
- Tinted glasses for photophobia
- Magnification devices, electronic low-vision aids
- Orientation and mobility training
Surgical / Device-Based
- Retinal implants (prostheses): Both epiretinal and subretinal microchip implants have been used with some success to improve light perception in very advanced RP (e.g., Argus II epiretinal prosthesis)
- Cataract extraction for posterior subcapsular cataracts when visually significant
- Intravitreal anti-VEGF or acetazolamide for cystoid macular edema
Gene Therapy (Emerging)
- RPE65 replacement (voretigene neparvovec / Luxturna): Approved for Leber congenital amaurosis and RPE65-associated RP; demonstrated improvement in visual function
- RPGR gene therapy (X-linked RP): A 2026 systematic review and meta-analysis (PMID: 41773775) evaluated the efficacy and safety of gene therapy for RPGR-associated X-linked RP - representing the frontier of treatment for this form
- Multiple clinical trials ongoing across various genetic subtypes
Prognosis
- Highly variable depending on inheritance pattern and specific gene mutation
- Autosomal dominant forms are typically milder and progress more slowly; patients may retain useful vision into middle or late adult life
- Autosomal recessive and X-linked forms tend to be more severe with earlier vision loss
- Most patients eventually develop tunnel vision (preserved central field only); total blindness can occur but is not universal
- Legal blindness occurs in a significant proportion, typically between the 5th and 7th decades
Summary Table
| Feature | Details |
|---|---|
| Pathology | Rod-cone dystrophy; apoptotic photoreceptor death |
| Prevalence | 1:3,000-1:5,000 |
| Key genes | RHO, RPGR, peripherin, USH2A, EYS (>100 loci) |
| Classic triad | Bone-spicule pigment, arteriolar attenuation, waxy disc pallor |
| Key symptom | Night blindness (first); tunnel vision (late) |
| Diagnostic test | ERG (extinguished), visual fields (ring scotoma) |
| Treatment | Vitamin A, gene therapy (select subtypes), retinal prostheses |
| Associations | Usher, Bardet-Biedl, Refsum, Kearns-Sayre, LCA |
Sources: Kanski's Clinical Ophthalmology 10th ed., Harrison's Principles of Internal Medicine 22E, Goldman-Cecil Medicine, Robbins Cotran & Kumar Pathologic Basis of Disease, Wills Eye Manual
Recent evidence note: A 2026 systematic review (PMID: 41773775) confirms gene therapy for RPGR-associated X-linked RP shows efficacy and safety signals, though larger trials are still needed. The RHO p.P23H variant (the most common cause of AD-RP in Western populations) has been characterized with updated prevalence estimates in a 2024 meta-analysis (PMID: 39278389).
This is a shared conversation. Sign in to Orris to start your own chat.