Platelets disorder
Reading File
Asking for Preferences
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Searching Images
platelet disorders thrombocytopenia petechiae blood smear
I now have comprehensive content from multiple authoritative sources. Let me compose the full overview.
Platelet Disorders — Medical Student Overview
Normal Platelet Physiology (Quick Recap)
Platelets (thrombocytes) are anucleate cell fragments derived from megakaryocytes. Normal count: 150,000–400,000/µL. They carry two key surface glycoprotein complexes:
- GP Ib/IX — receptor for von Willebrand factor (vWF); mediates adhesion
- GP IIb/IIIa — receptor for fibrinogen; mediates aggregation
Classification of Platelet Disorders
Platelet disorders fall into two broad categories:
| Category | Problem | Platelet Count |
|---|---|---|
| Quantitative | Too few (thrombocytopenia) or too many (thrombocytosis) | Abnormal |
| Qualitative | Normal count, but dysfunction | Usually normal |
1. THROMBOCYTOPENIA (Platelet count < 150,000/µL)
Clinical Bleeding Thresholds
- < 150,000 — thrombocytopenia by definition; minimal spontaneous bleeding
- 20,000–50,000 — increased risk of post-traumatic bleeding
- < 5,000–10,000 — risk of spontaneous bleeding (petechiae, ecchymoses, mucosal hemorrhage, CNS bleeds)
The bleeding pattern is superficial: petechiae, ecchymoses, mucous membrane hemorrhage (nosebleeds, gum bleeding, menorrhagia). This contrasts with coagulation factor deficiencies, which cause deep bleeds (hemarthrosis, hematomas). — Robbins & Kumar Basic Pathology
Causes of Thrombocytopenia
A. Decreased Platelet Production
- Generalized bone marrow failure: aplastic anemia, leukemia/marrow infiltration
- Selective: drugs (alcohol, thiazides, cytotoxic agents), infections (measles, HIV)
- Ineffective megakaryopoiesis: megaloblastic anemia, paroxysmal nocturnal hemoglobinuria (PNH)
B. Increased Platelet Destruction
| Mechanism | Example |
|---|---|
| Immune (autoimmune) | ITP, SLE |
| Immune (drug-induced) | Heparin (HIT), quinidine, sulfa |
| Immune (alloimmune) | Post-transfusion purpura, neonatal alloimmune |
| Non-immune (microangiopathic) | DIC, TTP, HUS |
| Infection-associated | HIV, EBV, CMV |
C. Sequestration
- Hypersplenism (the spleen normally sequesters ~30% of circulating platelets; in massive splenomegaly this rises to 80–90%)
D. Dilutional
- Massive transfusion (stored RBCs/FFP lack platelets)
Key Specific Disorders
Immune Thrombocytopenic Purpura (ITP)
- Autoantibodies (IgG) target platelet GP IIb/IIIa or GP Ib/IX → opsonized platelets destroyed by splenic macrophages
- Antibodies detectable in ~80% of cases
- Acute ITP: children, post-viral, self-limited
- Chronic ITP: women age 20–40; insidious onset (petechiae, easy bruising, epistaxis, gum bleeding)
- Bone marrow shows increased megakaryocytes (compensatory) — hallmark of destructive thrombocytopenia
- Treatment: immunosuppressants, IVIG, splenectomy (complete remission in >2/3 of patients)
Heparin-Induced Thrombocytopenia (HIT)
- Occurs in 3–5% of patients on unfractionated heparin after 1–2 weeks
- Mechanism: IgG antibodies bind platelet factor 4 (PF4)–heparin complex → immune complexes activate platelets via Fc receptors → platelet consumption AND thrombosis
- Paradoxically causes thrombosis (venous + arterial) despite low platelets — a high-yield exam point
- Treatment: stop heparin immediately; switch to direct thrombin inhibitors (argatroban, bivalirudin); low-molecular-weight heparin reduces (but does not eliminate) risk
Thrombotic Thrombocytopenic Purpura (TTP)
Classic pentad (mnemonic: FAT RN):
- Fever
- Anemia (microangiopathic hemolytic — MAHA)
- Thrombocytopenia
- Renal failure
- Neurologic symptoms (transient deficits)
- Pathogenesis: deficiency of ADAMTS13 (a metalloprotease that cleaves ultra-large vWF multimers). Without ADAMTS13, ultra-large vWF accumulates → platelet-rich microthrombi throughout the microcirculation
- Can be congenital (Upshaw-Schulman syndrome) or acquired (autoantibody against ADAMTS13)
- PT and aPTT are normal (unlike DIC) — key exam distinction
- Treatment: plasma exchange (replaces ADAMTS13 and removes antibodies)
Hemolytic Uremic Syndrome (HUS)
- Shares MAHA + thrombocytopenia with TTP
- Key difference: predominant acute renal failure; neurologic symptoms less prominent; more common in children
- Classic trigger: E. coli O157:H7 Shiga toxin → endothelial damage in renal microvasculature
- Complement dysregulation also plays a role
- PT/aPTT normal (unlike DIC)
TTP vs HUS vs DIC — Exam Table
| Feature | TTP | HUS | DIC |
|---|---|---|---|
| MAHA | ✓ | ✓ | ✓ |
| Thrombocytopenia | ✓ | ✓ | ✓ |
| Neurologic sx | Prominent | Rare | Variable |
| Renal failure | Mild | Severe | Variable |
| PT/aPTT | Normal | Normal | Prolonged |
| ADAMTS13 | ↓↓ | Normal | Normal |
| Fibrinogen | Normal | Normal | ↓ |
2. THROMBOCYTOSIS (Platelet count > 400,000/µL)
Reactive (Secondary) Thrombocytosis
- Most common cause
- Causes: iron deficiency, inflammation, infection, malignancy, splenectomy, tissue damage
- Platelets usually < 1,000,000/µL
- No increased thrombotic or bleeding risk — platelets are functionally normal
- Treat the underlying cause
Essential (Primary) Thrombocythemia (ET)
- Clonal myeloproliferative neoplasm (stem cell disorder)
- Platelet count often > 1,000,000/µL
- JAK2 V617F mutation present in ~50% of cases (confirms clonal origin; its absence does not rule out ET)
- Can cause both thrombosis AND bleeding
- Markedly elevated platelets (>1.5 million) can cause acquired von Willebrand disease (platelets bind and remove vWF from circulation) → paradoxical bleeding
- Treatment: hydroxyurea (cytoreduction), aspirin; anagrelide
3. QUALITATIVE PLATELET DISORDERS (Normal Count, Dysfunctional Platelets)
Inherited Disorders
| Disorder | Defect | Mechanism | Presentation |
|---|---|---|---|
| Bernard-Soulier Syndrome | ↓ GP Ib/IX | Cannot bind vWF → defective adhesion | Severe bleeding; giant platelets on smear |
| Glanzmann Thrombasthenia | ↓ GP IIb/IIIa | Cannot bind fibrinogen → defective aggregation | Severe bleeding; normal platelet count/size |
| Storage Pool Disease (δ-SPD) | ↓ dense granules (ADP/serotonin) | Defective release reaction | Mild-moderate bleeding |
| Hermansky-Pudlak Syndrome | ↓ δ-granules + albinism | Lysosomal trafficking defect | Bleeding + oculocutaneous albinism |
Both Bernard-Soulier and Glanzmann are autosomal recessive, present in childhood with severe bleeding. — Robbins & Kumar Pathologic Basis of Disease
Acquired Disorders
| Cause | Mechanism | Notes |
|---|---|---|
| Aspirin / NSAIDs | Irreversible inhibition of COX-1 → ↓ thromboxane A2 | Effect lasts platelet lifespan (~7–10 days) |
| Uremia | Multifactorial (↓ adhesion, secretion, aggregation) | Improved by dialysis, DDAVP, raising Hct to 27–32% |
| Cardiopulmonary bypass | Mechanical activation and exhaustion | Responds to platelet transfusion |
| Myeloproliferative/MDS | Intrinsic platelet defects, circulating paraproteins |
4. MORPHOLOGIC CLUES ON BLOOD SMEAR
| Finding | Diagnosis |
|---|---|
| Giant platelets | Bernard-Soulier, MYH9-related (May-Hegglin anomaly) |
| Small platelets | Wiskott-Aldrich syndrome |
| Agranular (gray) platelets | Gray platelet syndrome |
| Platelet aggregates | Pseudo-thrombocytopenia (EDTA artifact) — redraw in citrate |
| Schistocytes + low platelets | TTP, HUS, DIC (MAHA) |
| Platelet satellitism | EDTA-dependent artifact around neutrophils |
5. KEY DIAGNOSTIC LABS — Quick Reference
| Test | Purpose |
|---|---|
| CBC + peripheral smear | First step — always confirm automated counts with smear |
| PT/aPTT | Normal in ITP, TTP, HUS; prolonged in DIC |
| Fibrinogen + D-dimer | ↓ fibrinogen, ↑ D-dimer in DIC |
| ADAMTS13 activity | Confirms TTP when <10% |
| PF4-heparin antibody (ELISA + serotonin release assay) | HIT diagnosis |
| Platelet aggregation studies | Glanzmann (no aggregation with ADP/collagen), Bernard-Soulier (no ristocetin aggregation) |
| Bone marrow biopsy | ↑ megakaryocytes = destructive; ↓ = production failure |
| JAK2 V617F | Essential thrombocythemia / other MPNs |
Visual Reference
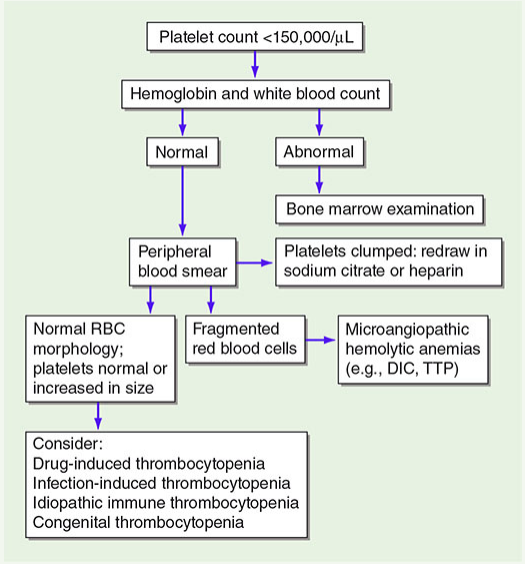
Thrombocytopenia Diagnostic Algorithm (Harrison's Principles):
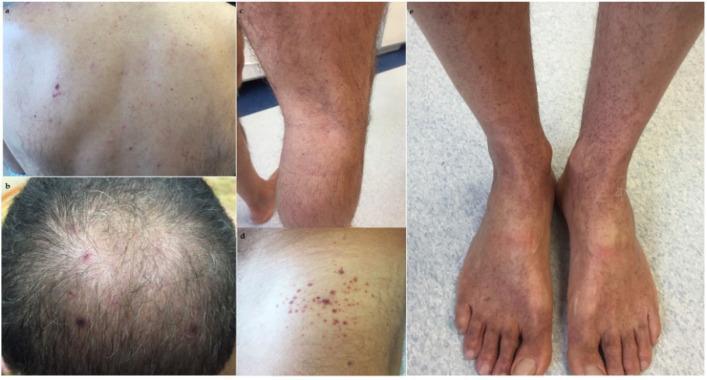
Clinical petechiae in ITP (lower extremity distribution):
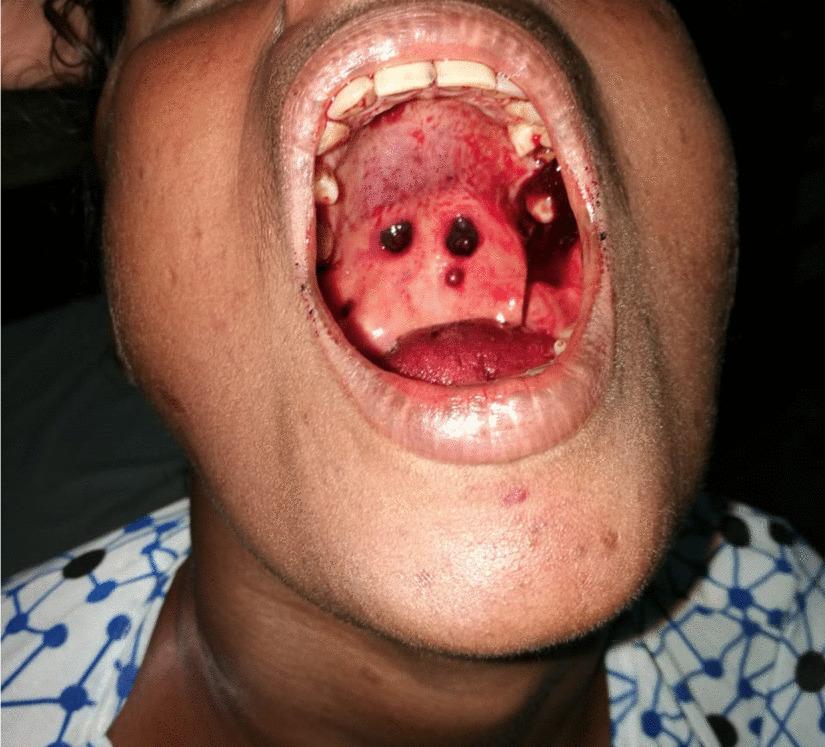
Oral wet purpura and petechiae in severe thrombocytopenia:
High-Yield Exam Mnemonics
- HIT = Heparin → IgG → PF4 → Thrombosis (counterintuitive: low platelets + clot)
- TTP = ADAMTS13 deficiency + MAHA + normal PT/PTT
- TTP vs HUS: TTP = brain; HUS = kidney (kids + E. coli)
- Glanzmann = aggregation defect (GP IIb/IIIa — "IIb/IIIa = aggrIIBregation")
- Bernard-Soulier = adhesion defect (GP Ib/IX — "Ib = adheIBsion")
- Aspirin effect: irreversible COX inhibition → lasts 7–10 days (entire platelet lifespan)
Sources: Robbins & Kumar Basic Pathology; Robbins, Cotran & Kumar Pathologic Basis of Disease; Harrison's Principles of Internal Medicine 22E (2025); Goldman-Cecil Medicine; Henry's Clinical Diagnosis and Management by Laboratory Methods
This is a shared conversation. Sign in to Orris to start your own chat.