Describe the etiology and pathogenis of avn hip from campbell book of Orthopaedics
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have all the information needed to write a comprehensive answer directly from Campbell's Operative Orthopaedics 15th Ed 2026. Here is the full response:
Osteonecrosis (AVN) of the Femoral Head - Etiology and Pathogenesis
Source: Campbell's Operative Orthopaedics, 15th Edition (2026)
Definition and Overview
Osteonecrosis (formerly called avascular necrosis) of the femoral head is a progressive disease that generally affects patients in the third through fifth decades of life. Simply defined, osteonecrosis means "dead bone." The "avascular" state of the necrotic bone results from a loss of circulation arising from numerous potential causes.
It is estimated that 20,000 new cases are diagnosed each year in the United States. When symptomatic, it typically leads to collapse of the femoral head and eventual deterioration of the hip joint.
Osteonecrosis describes an end condition that is the result of many possible pathogenic pathways. The process is most likely multifactorial - some patients with known risk factors never develop it, while many without identifiable risk factors do, suggesting individual susceptibility.
Etiology - Risk Factors (Box 7.4)
| Category | Specific Factors |
|---|---|
| Traumatic | Femoral head fractures, hip dislocation, surgery |
| Pharmacologic | Corticosteroid use |
| Toxic | Alcohol abuse, smoking |
| Hematologic | Hemoglobinopathies (sickle cell anemia), coagulation disorders, myeloproliferative disorders (Gaucher disease, leukemia) |
| Physical | Hyperbaric decompression (dysbarism) |
| Metabolic | Hyperlipidemias |
| Systemic disease | Chronic kidney disease, autoimmune diseases, HIV |
| Idiopathic | No identifiable cause (designated as idiopathic osteonecrosis) |
Pathogenesis - Mechanisms (Box 7.5)
Campbell's organizes the pathogenic mechanisms into two main categories:
1. Ischemia
This is the central final pathway - interruption of blood supply to the femoral head. It occurs through four sub-mechanisms:
A. Vascular Disruption (Mechanical)
- Femoral head fracture
- Hip dislocation
- Surgery
These directly tear or kink the retinacular vessels (mainly the medial femoral circumflex artery branches) supplying the femoral head.
B. Vascular Compression or Constriction
- Increased intraosseous pressure caused by marrow fatty infiltration
- Caused by: corticosteroids, alcohol
Corticosteroids and alcohol promote fat accumulation in marrow cells (adipogenesis). The swollen fat cells increase intraosseous pressure within the rigid bony compartment of the femoral head, compressing the sinusoidal vessels and reducing perfusion.
C. Vasoconstriction of Arteries Perfusing the Femoral Head
- Corticosteroids
- Endothelial nitric oxide synthase (eNOS) polymorphisms - reduced NO production leads to impaired vasodilation
D. Intravascular Occlusion
This is further subdivided into:
-
Thrombosis - due to a hypercoagulable/thrombophilic state:
- Low Protein C and Protein S
- Activated protein C resistance (Factor V Leiden mutation)
- Elevated homocysteine
- eNOS polymorphisms
- Hypofibrinolysis:
- High plasminogen activator inhibitor (PAI) activity
- PAI-1 polymorphisms
- High lipoprotein(a)
-
Embolization - fat emboli or air emboli occluding the microvasculature
-
Sickle cell occlusion - sickling of erythrocytes in the small vessels of the femoral head causes vascular stasis and obstruction
2. Direct Cellular Toxicity
Even without ischemia, some agents can kill osteocytes and osteoblasts directly:
- Pharmacologic agents (e.g., corticosteroids, alcohol)
- Irradiation - radiation-induced damage to cells and small vessels
- Oxidative stress - free radical injury to bone cells
- Altered differentiation of mesenchymal stem cells - corticosteroids and alcohol divert mesenchymal stem cells preferentially toward adipogenesis at the expense of osteogenesis, leading to defective bone repair
- Combined corticosteroid/alcohol toxicity
The Pathologic Sequence (How AVN Progresses to Collapse)
Although Campbell's lists these mechanisms, the final common pathway is:
- Loss of blood supply to a segment of the femoral head (typically the anterosuperior weight-bearing segment)
- Osteocyte death within 12-48 hours of ischemia
- Necrotic segment is structurally weakened - trabeculae lose viability
- Reactive interface forms at the margin of necrotic and viable bone (seen as the "reactive line" on MRI - the classic double-line sign)
- Subchondral fracture occurs along this interface under repetitive loading - this is the crescent sign on X-ray (pathognomonic for Stage III)
- Femoral head collapse - the necrotic segment loses structural support and caves in
- Secondary osteoarthritis - joint space loss and acetabular involvement follow
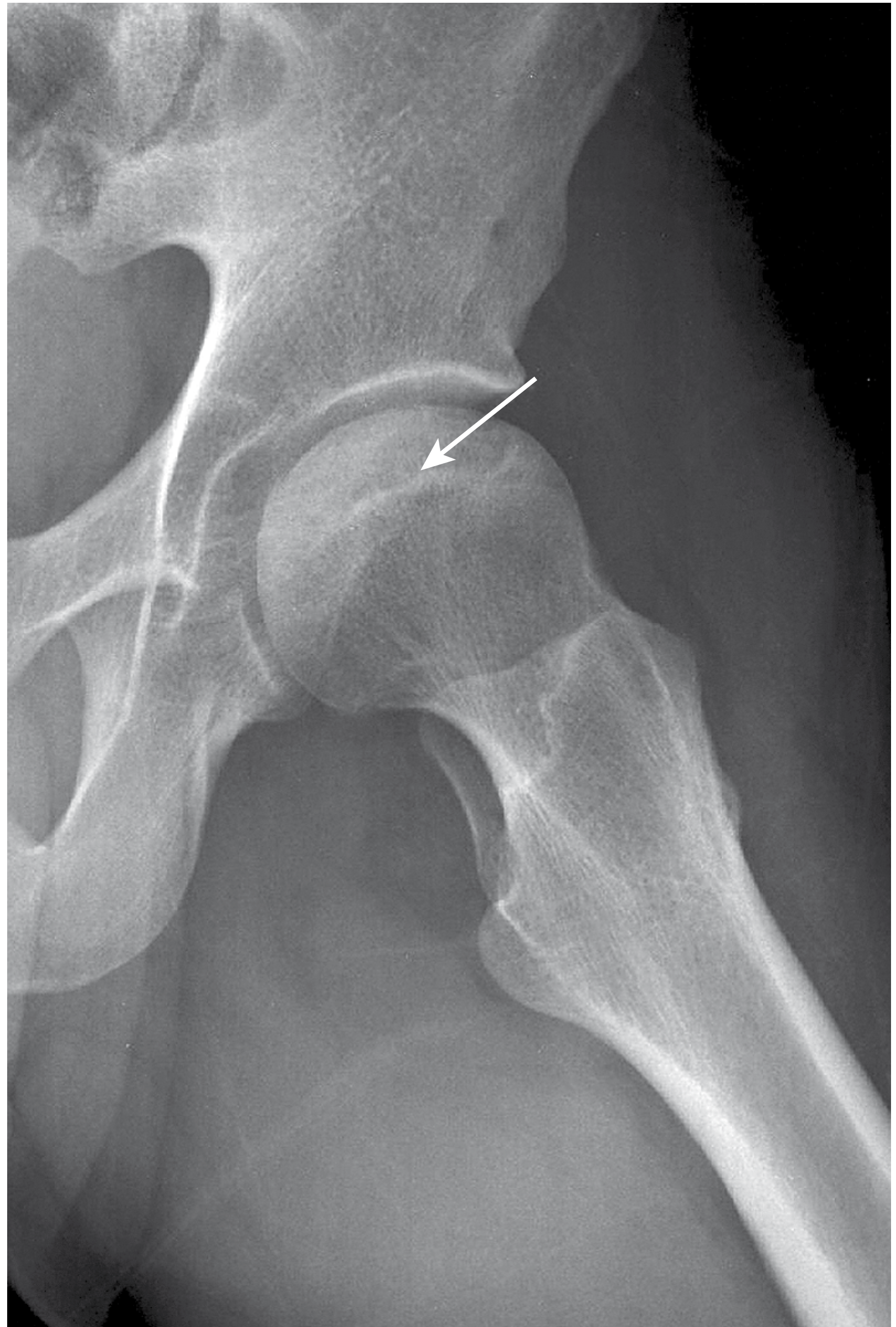
Figure 7.46 from Campbell's - Crescent sign in stage III osteonecrosis of the hip
Key Takeaways from Campbell's
- The most important risk factors are trauma (fracture/dislocation), corticosteroid use, and alcohol abuse
- The process is multifactorial - no single theory fully accounts for all causes
- The thrombophilia/hypofibrinolysis mechanism is particularly important for explaining non-traumatic idiopathic cases
- Corticosteroids act through multiple mechanisms simultaneously: marrow fat infiltration (compressive ischemia), vasoconstriction (eNOS), and direct cellular toxicity (altered MSC differentiation)
- The combined necrotic angle on MRI (midcoronal + midsagittal) predicts collapse: no collapse with <190°, collapse in all cases with >240°
Source: Campbell's Operative Orthopaedics, 15th Ed (2026), Chapter 7 - Osteonecrosis of the Femoral Head, Box 7.4 and Box 7.5 (modified from Zalavras CG, Lieberman JR, JAAOS 22:455, 2014)
Describe itcin a answer form for a final year ms orthopaedics exam
Here is a model exam answer for MS Orthopaedics finals:
Etiology and Pathogenesis of Avascular Necrosis (Osteonecrosis) of the Femoral Head
Introduction
Osteonecrosis (AVN) of the femoral head is a progressive condition resulting from interruption of blood supply to the femoral head, leading to bone death, subchondral collapse, and eventual secondary osteoarthritis. It predominantly affects patients in the 3rd to 5th decade of life. Approximately 20,000 new cases are diagnosed annually in the United States. The condition is the end result of multiple possible pathogenic pathways and is best considered multifactorial in origin.
Etiology
The causes are broadly classified as traumatic and non-traumatic (atraumatic).
A. Traumatic Causes
These directly disrupt the blood supply to the femoral head, particularly the terminal branches of the medial femoral circumflex artery (MFCA).
- Displaced femoral neck fractures - the most common traumatic cause; the retinacular vessels are torn
- Hip dislocation - posterior dislocation especially; kinks and stretches the capsular vessels
- Surgical trauma - reduction manoeuvres, extensive capsulotomy
The risk of AVN after displaced femoral neck fracture is 15-50% and after posterior hip dislocation is 10-25%.
B. Atraumatic (Non-traumatic) Causes
| Risk Factor | Mechanism |
|---|---|
| Corticosteroid use | Marrow fat infiltration → increased intraosseous pressure; vasoconstriction via eNOS suppression; direct osteocyte toxicity |
| Alcohol abuse | Fat emboli to femoral head vessels; increased intraosseous pressure; direct cellular toxicity |
| Sickle cell disease | Sickling of RBCs → vascular stasis and occlusion in sinusoidal vessels |
| Smoking | Vasoconstriction, impaired fibrinolysis |
| Hyperlipidemias | Fat emboli, intravascular lipid deposition |
| Coagulation disorders | Thrombophilia (low Protein C & S, Factor V Leiden, high homocysteine) → intravascular thrombosis |
| Dysbarism (decompression sickness) | Nitrogen gas emboli occlude vessels |
| Gaucher disease / Leukemia | Marrow infiltration → raised intraosseous pressure |
| Chronic kidney disease | Altered lipid and calcium metabolism |
| Autoimmune diseases / SLE | Often steroid-related; immune complex vasculitis |
| HIV | Direct effect + highly active antiretroviral therapy (HAART) related lipid abnormalities |
| Idiopathic | No identifiable cause; individual genetic susceptibility likely |
Exam point: Corticosteroids and alcohol together account for over 90% of non-traumatic AVN cases.
Pathogenesis
Campbell's organizes the pathogenic mechanisms into two broad categories:
Category 1 - Ischemia
This is the central and final common pathway in almost all forms of AVN. Ischemia is produced by four distinct mechanisms:
1. Vascular Disruption
Direct mechanical disruption of vessels (fracture, dislocation, surgery). The lateral epiphyseal branches of the MFCA, which run along the posterior capsule as retinacular vessels, are the most vulnerable. Disruption leads to immediate cessation of blood flow.
2. Vascular Compression / Raised Intraosseous Pressure
- Corticosteroids and alcohol both stimulate marrow adipogenesis - conversion of haematopoietic marrow to fat cells
- Fat cells enlarge within the rigid intraosseous compartment of the femoral head
- This raises intraosseous pressure, compressing sinusoidal vessels
- Normal intraosseous pressure: ~30 mmHg; in AVN it may exceed 50 mmHg
- Sinusoidal flow ceases → ischemia
3. Vasoconstriction
- Corticosteroids suppress endothelial nitric oxide synthase (eNOS)
- Reduced NO production → impaired vasodilation of arteries supplying the femoral head
- eNOS polymorphisms create individual susceptibility even without steroid use
4. Intravascular Occlusion
This occurs through two sub-mechanisms:
(a) Thrombosis / Thrombophilia:
- Low Protein C and Protein S
- Activated Protein C resistance (Factor V Leiden mutation)
- Elevated homocysteine
- eNOS polymorphisms
(b) Hypofibrinolysis:
- Elevated plasminogen activator inhibitor (PAI) activity
- PAI-1 polymorphisms
- Elevated lipoprotein(a) These states prevent clot lysis in the microcirculation of the femoral head.
(c) Embolization:
- Fat emboli (from lipid-laden marrow, alcohol use, fractures)
- Air emboli (decompression sickness / dysbarism)
(d) Sickle cell occlusion:
- Deoxygenated HbS polymerizes → rigid sickled cells lodge in sinusoidal vessels → mechanical occlusion → ischemia
Category 2 - Direct Cellular Toxicity
In addition to ischemia, some agents kill osteocytes and bone cells directly, independent of vascular events:
- Pharmacologic agents - corticosteroids and alcohol act directly on osteocytes and marrow stromal cells
- Irradiation - radiation causes direct DNA damage and kills osteoblasts; also damages small vessels (radiation arteritis)
- Oxidative stress - reactive oxygen species generated by alcohol metabolism damage bone cell membranes
- Altered mesenchymal stem cell (MSC) differentiation - both corticosteroids and alcohol shift MSC differentiation toward adipogenesis and away from osteogenesis, resulting in defective repair of necrotic bone even when vascularity is partially restored
- Combined corticosteroid and alcohol toxicity
The Pathological Sequence - How AVN Progresses to Collapse
Understanding the progression is essential for staging and management:
Blood supply interrupted
↓
Osteocyte death (within 12-48 hours of ischemia)
↓
Necrotic segment in anterosuperior femoral head
↓
Reactive interface forms between viable and necrotic bone
(seen as "double line sign" on MRI - pathognomonic)
↓
Fibrovascular repair tissue attempts healing from periphery
(creeping substitution - inadequate in weight-bearing bone)
↓
Repeated mechanical loading on structurally dead trabeculae
↓
Subchondral fracture → CRESCENT SIGN on X-ray (Stage III)
↓
Femoral head collapse and flattening
↓
Secondary osteoarthritis (Stage IV/V)
The anterosuperior quadrant of the femoral head is the most commonly involved because it is the principal weight-bearing area and has the most tenuous blood supply (end-arterial territory of the lateral epiphyseal vessels).
Why the Femoral Head is Particularly Vulnerable
- End-arterial supply - the lateral epiphyseal branches of the MFCA are end arteries with minimal collateral circulation
- Intracapsular location - capsular tamponade from haemarthrosis further compromises already tenuous vessels after injury
- High mechanical demand - the femoral head bears 3-4x body weight during walking, accelerating collapse of necrotic trabeculae
- Rigid compartment - the bony cortex prevents pressure decompression when intraosseous pressure rises
Summary
| Mechanism | Examples | Final Effect |
|---|---|---|
| Vascular disruption | Fracture, dislocation | Acute ischemia |
| Raised intraosseous pressure | Steroids, alcohol (fat infiltration) | Compressive ischemia |
| Vasoconstriction | Steroids (eNOS suppression) | Reduced perfusion |
| Intravascular occlusion | Thrombophilia, sickle cell, emboli | Microvascular ischemia |
| Direct cellular toxicity | Steroids, alcohol, radiation | Osteocyte death without ischemia |
All pathways ultimately lead to osteocyte death → failed repair → subchondral fracture → collapse.
Based on Campbell's Operative Orthopaedics, 15th Edition (2026), Chapter 7
Describe etiologies in detail
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
I now have sufficient detail from Campbell's across multiple sections. Here is the comprehensive exam answer:
Etiology of Avascular Necrosis (Osteonecrosis) of the Femoral Head - Detailed Description
Based on Campbell's Operative Orthopaedics, 15th Edition (2026)
Classification of Etiology
The causes of AVN are broadly divided into traumatic and atraumatic (non-traumatic) categories. No single theory fully explains all cases - the process is most likely multifactorial, and individual genetic susceptibility plays a role (some patients with risk factors never develop AVN; others without any identifiable factor do).
I. TRAUMATIC CAUSES
1. Displaced Femoral Neck Fracture
- The single most important traumatic cause
- The retinacular vessels (lateral epiphyseal branches of the medial femoral circumflex artery, MFCA) run along the posterior femoral neck within the capsule; displaced fractures directly tear or kink these vessels
- The intracapsular haematoma further raises capsular pressure, compressing the already-disrupted vessels
- Risk of AVN: 15-50% for displaced fractures (Garden III/IV); much lower for undisplaced fractures
- The risk increases with: degree of displacement, time to reduction (>6-12 hours significantly increases risk), and quality of reduction
2. Traumatic Hip Dislocation
- Posterior dislocation is the most common and most dangerous
- The MFCA branches are stretched, kinked, or torn as the femoral head is levered out posteriorly
- Risk of AVN: 10-25% after posterior dislocation
- Risk increases with: delay to reduction (>6 hours dramatically increases risk), severity of initial displacement, and associated acetabular fractures
- Anterior dislocation (rare) can also cause AVN by direct pressure on the anterior vessels
3. Surgical Trauma
- Extensive capsulotomy, prolonged retraction, or inadvertent vessel injury during hip surgery can cause AVN
- Valgus osteotomy of the proximal femur can compromise the remaining blood supply if vessels are already marginal
II. ATRAUMATIC (NON-TRAUMATIC) CAUSES
1. Corticosteroid Use (Most Common Atraumatic Cause)
Corticosteroids are responsible for approximately 35-40% of all atraumatic AVN cases. They act through multiple simultaneous mechanisms:
(a) Marrow fat infiltration and raised intraosseous pressure:
- Steroids strongly promote adipogenesis - differentiation of mesenchymal stem cells (MSCs) into fat cells rather than osteoblasts
- Fat cell hypertrophy within the rigid intraosseous compartment raises intraosseous pressure
- This compresses sinusoidal vessels and impairs venous outflow, leading to ischemia
(b) Vasoconstriction:
- Steroids suppress endothelial nitric oxide synthase (eNOS)
- Reduced NO production → impaired arterial vasodilation → reduced perfusion pressure to the femoral head
(c) Intravascular fat emboli:
- Steroid-induced hyperlipidaemia promotes fat emboli that occlude microvessels in the femoral head
(d) Direct cellular toxicity:
- Direct toxic effect on osteocytes and osteoblasts, impairing their viability and repair capacity
- Suppresses bone formation while increasing bone resorption
(e) Altered MSC differentiation:
- Shifts stem cell lineage away from osteogenesis toward adipogenesis, impairing the repair response to any ischemic insult
Dose and duration - risk increases with high-dose (>2 g prednisolone equivalent in first year) and prolonged use. Common clinical contexts: organ transplantation, SLE, asthma, inflammatory bowel disease, chemotherapy protocols.
Exam point: Corticosteroid AVN is often bilateral (up to 72% of cases) and affects the femoral head most frequently, followed by the humeral head, femoral condyle, and talus.
2. Alcohol Abuse (Second Most Common Atraumatic Cause)
Accounts for approximately 20-30% of atraumatic AVN. Mechanisms include:
(a) Fat emboli:
- Alcohol causes hyperlipidaemia and promotes release of fat from liver and marrow stores
- Fat emboli from these sources occlude the small sinusoidal vessels of the femoral head
(b) Raised intraosseous pressure:
- Alcohol stimulates marrow adipogenesis (similar to steroids), increasing fat cell volume within the femoral head
- This raises intraosseous pressure and compresses sinusoidal channels
(c) Direct hepatotoxicity and coagulopathy:
- Alcohol-induced liver disease impairs synthesis of coagulation factors and natural anticoagulants (Protein C, Protein S)
- This creates a prothrombotic state that predisposes to microvascular thrombosis in the femoral head
(d) Direct cellular toxicity:
- Alcohol metabolites (acetaldehyde) are directly toxic to osteocytes
- Oxidative stress from alcohol metabolism generates reactive oxygen species (ROS) that damage bone cell membranes
- Suppresses osteoblast function and bone repair capacity
(e) Altered MSC differentiation:
- Like steroids, alcohol shifts stem cell fate toward adipogenesis
Dose relationship: Risk increases with chronic heavy use (>400 ml ethanol/week). The risk is not related to a single episode of bingeing but to chronic cumulative exposure.
3. Sickle Cell Disease (Haemoglobinopathy)
- AVN affects up to 50% of patients with sickle cell anaemia (HbSS) by adulthood
- Also occurs in HbSC disease and sickle cell trait (less frequently)
- Process can be bilateral in nearly 50% of affected patients
Mechanism:
- Deoxygenation triggers polymerization of HbS → rigid, sickle-shaped RBCs
- These cannot deform to pass through narrow sinusoidal channels of the femoral head
- Mechanical vascular occlusion → stasis → thrombosis → ischemia
- Repeated vaso-occlusive crises cause progressive cumulative ischemic damage
- The femoral head, with its slow-flow sinusoidal circulation and high oxygen demand, is particularly vulnerable
4. Thrombophilia and Coagulation Disorders
A group of inherited or acquired hypercoagulable states that cause microvascular thrombosis in the femoral head sinusoids:
| Condition | Mechanism |
|---|---|
| Low Protein C and Protein S | Failure to inhibit activated Factor Va and VIIIa → uncontrolled thrombin generation → thrombosis |
| Activated Protein C resistance (Factor V Leiden mutation) | Factor V cannot be inactivated → persistent procoagulant state |
| Elevated homocysteine (hyperhomocysteinaemia) | Endothelial damage + prothrombotic effect on coagulation cascade |
| eNOS polymorphisms | Reduced nitric oxide → vasoconstriction + prothrombotic endothelial state |
Hypofibrinolysis (failure to dissolve clots once formed):
- High Plasminogen Activator Inhibitor (PAI) activity - inhibits tissue plasminogen activator (tPA), preventing fibrinolysis
- PAI-1 polymorphisms - genetic variants that constitutively elevate PAI levels
- Elevated Lipoprotein(a) - competes with plasminogen for fibrin binding sites, impairing fibrinolysis
These coagulation abnormalities are particularly important in explaining idiopathic AVN - patients with no obvious risk factor but a cryptic thrombophilic tendency.
5. Hyperlipidaemia
- Mechanism: Elevated serum lipids → fat emboli lodge in sinusoidal vessels of the femoral head
- Lipid deposits in vessel walls also cause direct endothelial damage
- Seen in primary hypercholesterolaemia and as a secondary effect of steroids, alcohol, chronic kidney disease
- Statin use has been shown to have a protective effect in steroid-treated patients (by reducing lipid levels and possibly through pleiotropic effects on endothelium)
6. Dysbarism (Caisson Disease / Decompression Sickness)
- Affects deep-sea divers, tunnel workers (caisson workers), and aviators
- At high pressures, nitrogen dissolves into body fluids and tissues
- With rapid decompression (surfacing too quickly), dissolved nitrogen comes out of solution as gas bubbles
- These nitrogen gas bubbles act as emboli that occlude the microcirculation of the femoral head and other bones
- Also causes direct physical disruption of marrow cells
- AVN from dysbarism characteristically affects multiple bones and both femoral heads simultaneously
- Prevention: staged decompression protocols
7. Gaucher Disease (Myeloproliferative / Storage Disorder)
- An autosomal recessive lysosomal storage disorder - deficiency of glucocerebrosidase enzyme
- Glucocerebroside accumulates in macrophages (Gaucher cells) in bone marrow, liver, and spleen
- Mechanism of AVN: Bone marrow is progressively replaced by Gaucher cells → marrow packing → raised intraosseous pressure → sinusoidal compression → ischemia and infarction
- The medullary canal is widened, bone quality is poor (moth-eaten trabeculae, patchy sclerosis), making surgical fixation technically challenging
- Enzyme replacement therapy (imiglucerase) can partially ameliorate osseous complications
8. Radiation
- Radiation causes direct DNA damage to osteocytes and osteoblasts, leading to cell death
- Also causes radiation arteritis - progressive obliteration of small vessels supplying the femoral head
- AVN typically manifests months to years after radiation to the pelvis or hip (e.g., for gynaecological, bladder, or rectal malignancies)
- Dose-dependent: risk is significant above 50 Gy to the femoral head
9. Chronic Kidney Disease
- Altered calcium-phosphate metabolism causes secondary hyperparathyroidism and bone disease
- Renal transplant patients are doubly at risk - the disease itself and the post-transplant corticosteroid immunosuppression
- Uraemia may also cause a coagulopathy and vascular disease contributing to ischemia
10. Autoimmune Diseases (SLE, Rheumatoid Arthritis)
- Systemic Lupus Erythematosus (SLE) is one of the most common conditions associated with AVN
- The risk in SLE is partly from the high-dose steroid treatment used for flares, but also from:
- Antiphospholipid antibodies (lupus anticoagulant) → thrombophilia and microvascular thrombosis
- Immune complex deposition in vessel walls → vasculitis → ischemia
- AVN in SLE patients is often multifocal (femoral head + humeral head + other sites simultaneously)
11. Human Immunodeficiency Virus (HIV)
- AVN is significantly more common in HIV-positive patients
- Mechanisms include:
- Direct HIV-related vasculopathy
- Antiphospholipid antibodies are common in HIV (up to 50% of patients)
- Highly Active Antiretroviral Therapy (HAART) - particularly protease inhibitors cause lipodystrophy and hyperlipidaemia, promoting fat emboli and vascular occlusion
- Steroid use for HIV-related complications (PCP pneumonia, immune reconstitution)
12. Idiopathic (No Identifiable Cause)
- A significant proportion of AVN patients have no identifiable risk factor
- These are classified as idiopathic osteonecrosis
- Individual genetic susceptibility is likely - variants in genes regulating lipid metabolism, coagulation, MSC differentiation, and vascular tone (eNOS, PAI-1, Factor V) may create subclinical predisposition
- Up to 75% of patients with atraumatic osteonecrosis (including idiopathic) have radiographic or MRI evidence of bilateral hip disease at presentation, emphasising the systemic nature of the underlying condition
Summary Table
| Aetiology | Primary Mechanism | Key Clinical Context |
|---|---|---|
| Trauma (fracture/dislocation) | Vascular disruption | Garden III/IV NOF fracture; posterior dislocation |
| Corticosteroids | Fat infiltration, vasoconstriction, eNOS suppression, direct toxicity | Transplant, SLE, asthma |
| Alcohol | Fat emboli, direct toxicity, coagulopathy | Chronic heavy use >400 ml/week |
| Sickle cell disease | Mechanical RBC occlusion of sinusoids | HbSS, bilateral in 50% |
| Thrombophilia | Intravascular thrombosis, hypofibrinolysis | Low Prot C/S, Factor V Leiden, PAI-1 |
| Dysbarism | Nitrogen gas emboli | Divers, caisson workers |
| Gaucher disease | Marrow packing, raised intraosseous pressure | Young adults, splenomegaly, pancytopenia |
| Radiation | Direct osteocyte death, radiation arteritis | Post-pelvic radiotherapy |
| Hyperlipidaemia | Fat emboli, endothelial damage | Often co-exists with steroids/alcohol |
| HIV/HAART | Lipodystrophy, antiphospholipid Ab, vasculopathy | CD4 count decline + protease inhibitors |
| SLE | Antiphospholipid Ab, vasculitis, steroids | Multifocal AVN |
| CKD | Metabolic bone disease, steroid use post-transplant | Renal transplant recipients |
| Idiopathic | Genetic susceptibility, subclinical coagulopathy | 75% bilateral on MRI at presentation |
Source: Campbell's Operative Orthopaedics, 15th Edition (2026) - Chapter 7, Box 7.4 and Box 7.5; supplemented from Chapter 6 (Metabolic Disorders - Gaucher Disease and Sickle Cell)
This is a shared conversation. Sign in to Orris to start your own chat.