Memory
Reading File
Asking for Preferences
Finding Sources
Finding Sources
Searching the Web
neuroscience of memory formation hippocampus synaptic plasticity LTP 2025
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
I now have everything I need for a thorough, well-illustrated answer. Let me compile it.
The Neuroscience and Physiology of Memory
What Is Memory?
Memory is "the process by which knowledge is encoded, stored, and later retrieved." It is not a single function located in one brain region - different memory types depend on distinct neural circuits. Three major insights from decades of research:
- There are several forms of memory, each with different cognitive properties and brain substrates.
- Memory involves four operations: encoding, storage, consolidation, and retrieval.
- Imperfections in memory (amnesia, confabulation) reveal how normal memory works.
- Eric Kandel, Principles of Neural Science, 6th ed.
Taxonomy of Memory
Memory is classified along two main axes: time course and type of content.
By Time Course
| Stage | Clinical Term | Neuroscience Term | Duration | Anatomy |
|---|---|---|---|---|
| 1 | Immediate memory | Working memory | Seconds to ~20 sec without rehearsal | Prefrontal cortex, striatum |
| 2 | Short-term / recent memory | Episodic (recent) | Minutes to hours | Hippocampus + medial temporal lobe |
| 3 | Long-term / remote memory | Episodic (remote) + semantic | Weeks to lifetime | Neocortex (less hippocampus over time) |
Working memory holds roughly 5-9 meaningful items in conscious awareness. Without rehearsal, it fades in approximately 18-20 seconds. It relies primarily on prefrontal cortex (dorsolateral for spatial, ventrolateral for objects), with dopaminergic input from the midbrain and striatal modulation.
- Bradley and Daroff's Neurology in Clinical Practice
By Content: Explicit vs. Implicit
Explicit (declarative) memory - Conscious, can be verbalized. Damaged by medial temporal lobe lesions.
- Episodic memory: personal events ("what I did last Tuesday") - requires "mental time travel"
- Semantic memory: factual world knowledge ("Paris is the capital of France") - distributed across lateral temporal and association cortices
Implicit (non-declarative) memory - Unconscious, not verbalized. Survives medial temporal lobe damage.
- Procedural memory (skills and habits): processed in the striatum
- Classical/operant conditioning: amygdala (emotional responses), cerebellum (motor responses)
- Priming: improved recall from prior exposure; survives even in profound amnesia
- Habituation/sensitization: depends on basic reflex pathways
The landmark case of patient H.M. (bilateral hippocampal resection) showed that he could learn new motor skills (mirror drawing) and improve with priming tasks, but had no conscious recollection of having done them - directly proving the explicit/implicit dissociation.
- Kandel, Principles of Neural Science, pp. 1341-1342
Brain Structures: The Medial Temporal Lobe System
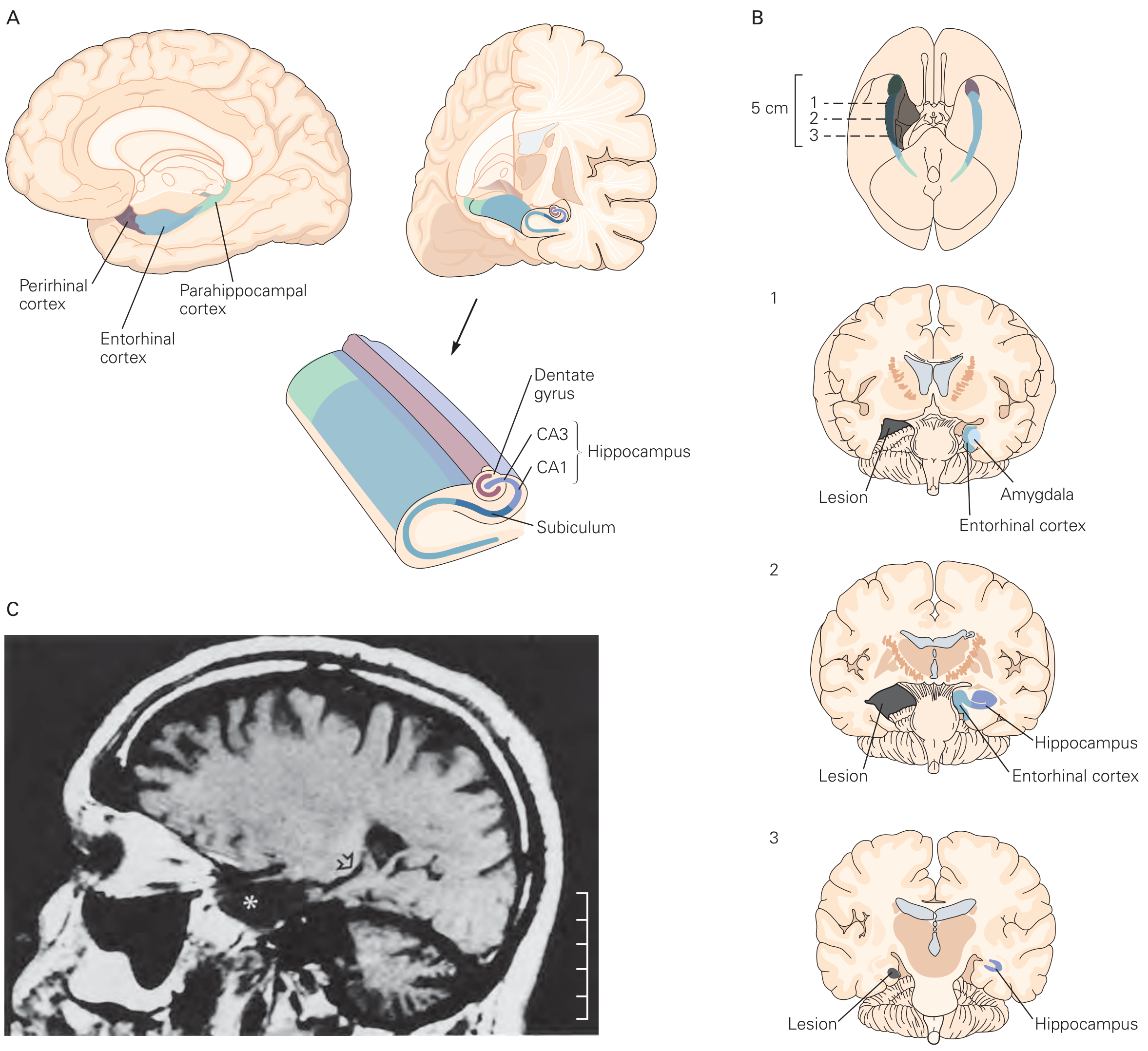
Figure: The medial temporal lobe (A), showing the hippocampus (CA1, CA3, dentate gyrus, subiculum), entorhinal cortex, perirhinal cortex, and parahippocampal cortex. Panel B shows H.M.'s bilateral lesion extent across 3 coronal levels. Panel C is H.M.'s actual MRI scan.
The hippocampal formation is the core structure for forming new episodic memories. It builds relational associations - linking objects, places, times, and people into a coherent episode. The hippocampus is NOT the final storage site; it acts as a temporary binding hub while memories are being consolidated to the neocortex.
A useful anatomical analogy (from Bradley's Neurology):
- Frontal lobes = the "filing clerk" that decides what to retrieve and from where
- Medial temporal lobes = the "recent memory filing cabinet" where new memories are stored
- Subcortical white matter = the pathway the filing clerk must travel to reach the cabinet
| Lesion site | Effect on memory |
|---|---|
| Medial temporal lobe (e.g., Alzheimer disease) | Damaged file cabinet - memories cannot be stored |
| Frontal lobe (e.g., stroke, tumor) | Disorganized filing clerk - poor organization and retrieval |
| Subcortical white matter (e.g., MS, ischemia) | Blocked pathway - slow/impaired access, intact on recognition testing |
Consolidation: From Short-Term to Long-Term
Consolidation occurs at two levels:
Synaptic Consolidation (Hours)
Driven by long-term potentiation (LTP) - a durable increase in synaptic transmission efficiency after repeated stimulation. LTP proceeds in two phases:
- Early LTP (E-LTP): protein synthesis-independent; lasts minutes to hours. The synapse becomes "tagged" via a protein synthesis-independent mechanism.
- Late LTP (L-LTP): requires intracellular signaling cascades and new protein synthesis in soma and dendrites; lasts days to years. The tagged synapse must capture plasticity-related proteins (PRPs) to stabilize.
The molecular trigger for most LTP:
- High-frequency presynaptic activity (or theta-burst stimulation, 4-8 Hz) depolarizes the postsynaptic membrane
- NMDA receptors (glutamate-gated, voltage-dependent) become unblocked - Na⁺ and Ca²⁺ flow in
- Calcium rise activates CaMKII (calcium-calmodulin-dependent kinase II), which autophosphorylates
- Cytoskeletal reconfiguration and AMPA receptor insertion strengthen the synapse
- Gene activation and new protein synthesis produce structural synaptic changes (dendritic spine growth)
"The key to memory is alteration in the strength of selected synaptic connections... the alteration involves the synthesis of proteins and the activation of genes. This occurs during the change from short-term working memory to long-term memory."
- Ganong's Review of Medical Physiology, 26th ed.
Systems Consolidation (Weeks to Years)
Over time, repeated reactivation of hippocampal-neocortical circuits gradually transfers memory representations to the neocortex. Once fully consolidated there, retrieval no longer requires the hippocampus - which is why patients with hippocampal damage (H.M., early Alzheimer disease) can recall childhood events (stored in neocortex) but not recent ones (not yet transferred).
Reconsolidation: Memory Is Not Fixed
A retrieved memory becomes transiently plastic again - a window called the reconsolidation window. After retrieval, the memory must re-stabilize through protein synthesis-dependent mechanisms similar to initial consolidation. This means:
- Existing memories can be strengthened or updated during reconsolidation
- Fear memories can be weakened or erased by pharmacologic or behavioral interventions during this window
This has generated significant clinical interest for PTSD treatment.
- Miller's Anesthesia, 10th ed.
The Amygdala and Emotional Memory
The amygdala (specifically the basolateral nucleus, BLA) is not essential for standard episodic memory, but it modulates the encoding and consolidation of emotionally significant events in the hippocampus via noradrenergic mechanisms. This explains why emotionally charged events are remembered more vividly and durably - a process that can become maladaptive in conditions like PTSD.
Summary Table: Brain Regions and Memory Functions
| Brain Region | Memory Role |
|---|---|
| Hippocampus (CA1, CA3, dentate gyrus) | New episodic memory encoding; relational binding; consolidation hub |
| Entorhinal/perirhinal/parahippocampal cortex | Gateway to hippocampus; object/context representation |
| Prefrontal cortex (dorsolateral/ventrolateral) | Working memory; retrieval strategy; executive control |
| Amygdala (basolateral nucleus) | Emotional memory modulation; fear conditioning |
| Striatum (basal ganglia) | Procedural memory; habit learning |
| Cerebellum | Motor conditioning (e.g., eyeblink conditioning) |
| Neocortex (association areas) | Long-term semantic and remote episodic storage |
Sources: Eric Kandel, Principles of Neural Science (6th ed.); Bradley and Daroff's Neurology in Clinical Practice; Ganong's Review of Medical Physiology (26th ed.); Miller's Anesthesia (10th ed.); Synaptic plasticity and memory hypothesis (PMC)
For a visual walkthrough of LTP and memory formation:
Synaptic Plasticity
Finding Sources
Finding Sources
Searching the Web
synaptic plasticity LTP LTD molecular mechanisms neuroscience 2025
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
I have comprehensive content. Now I'll compile the full answer.
Synaptic Plasticity
What It Is
Synaptic plasticity is the activity-dependent ability of synapses to change their strength - either strengthening or weakening - in response to patterns of use. It is the primary neurochemical foundation of learning and memory, and it also maintains network stability and governs how the brain adapts to experience.
"Changes in the strength and efficiency of synaptic signaling, termed synaptic plasticity, underlie one of the most important neurochemical foundations of learning and memory."
- Kaplan & Sadock's Comprehensive Textbook of Psychiatry
Overview: Three Major Categories
| Category | Direction | Duration | Function |
|---|---|---|---|
| Short-term plasticity | Up (facilitation) or down (depression) | Seconds to minutes | Filters and shapes signal transmission |
| Long-term potentiation (LTP) | Strengthening | Hours to years | Memory encoding and storage |
| Long-term depression (LTD) | Weakening | Hours to years | Memory refinement, forgetting, motor learning |
Plus two regulatory forms: homeostatic plasticity (network-wide gain control) and metaplasticity (plasticity of plasticity itself).
Short-Term Synaptic Plasticity
Short-term plasticity depends on the release probability (P) of a synapse:
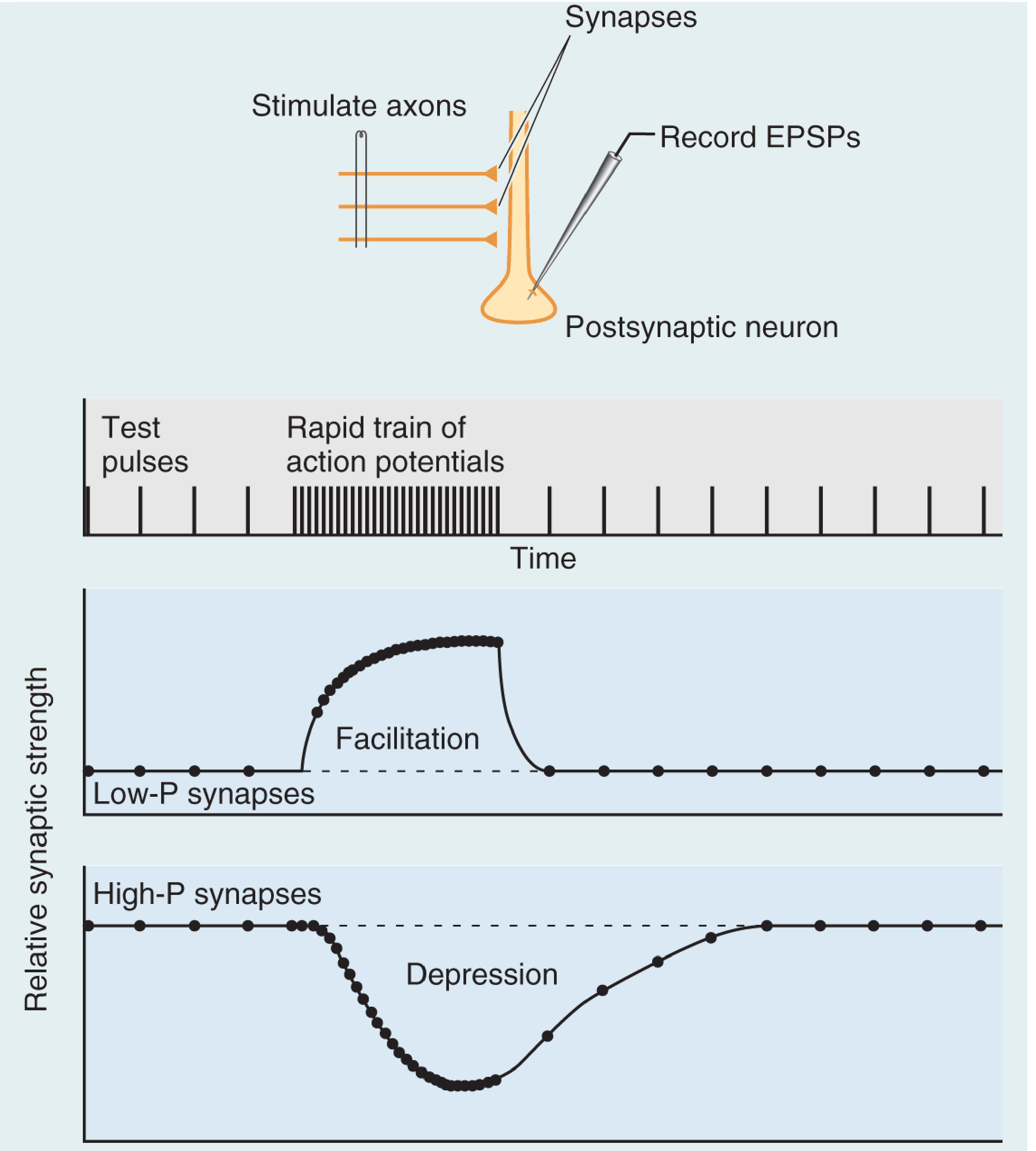
Figure: Repetitive stimulation of a presynaptic axon produces facilitation at low-P synapses (Ca²⁺ builds up, raising release probability) and depression at high-P synapses (vesicle pool depleted). Both reset to baseline after stimulation ends. From Neuroscience: Exploring the Brain, 5th ed.
Facilitation (low-P synapses): Infrequent spikes are unreliable, but rapid bursts cause Ca²⁺ to accumulate in the axon terminal before it can be cleared - making release virtually assured. These synapses are specialized to filter low-frequency signals while faithfully transmitting high-frequency bursts.
Depression (high-P synapses): Because they release reliably on each spike, the vesicle pool depletes during rapid trains. Transmission recovers only when vesicles are replenished from the reserve pool.
- Neuroscience: Exploring the Brain, 5th ed.
Long-Term Potentiation (LTP)
LTP is "a durable increase in synaptic transmission efficiency following a stimulation protocol" that persists from hours to years and is the dominant cellular model for memory storage.
The Hebbian Principle
Donald Hebb proposed that a synapse strengthens when it successfully participates in firing the postsynaptic neuron - "neurons that fire together, wire together." LTP is the biophysical implementation of this idea.
Three Key Properties (Kandel)
LTP at NMDA-receptor-dependent synapses (e.g. hippocampal CA1) has three properties that make it ideal for information storage:
-
Cooperativity - A single weak input cannot induce LTP (can't expel Mg²⁺ from NMDA channel). Only convergent activation of many inputs simultaneously achieves the strong depolarization required. This ensures only significant events trigger memory formation.
-
Associativity - A weak input paired with a strong one achieves LTP in both, because the strong input provides the depolarization. This is the cellular analog of Pavlovian conditioning - a neutral stimulus gains meaning when paired with a meaningful one.
-
Synapse specificity - Only activated synapses undergo LTP, even when neighboring synapses on the same cell receive strong stimulation. This allows a single neuron to store vast amounts of independent information across its thousands of synapses.
- Kandel, Principles of Neural Science, 6th ed., p. 1397
Molecular Mechanism of LTP
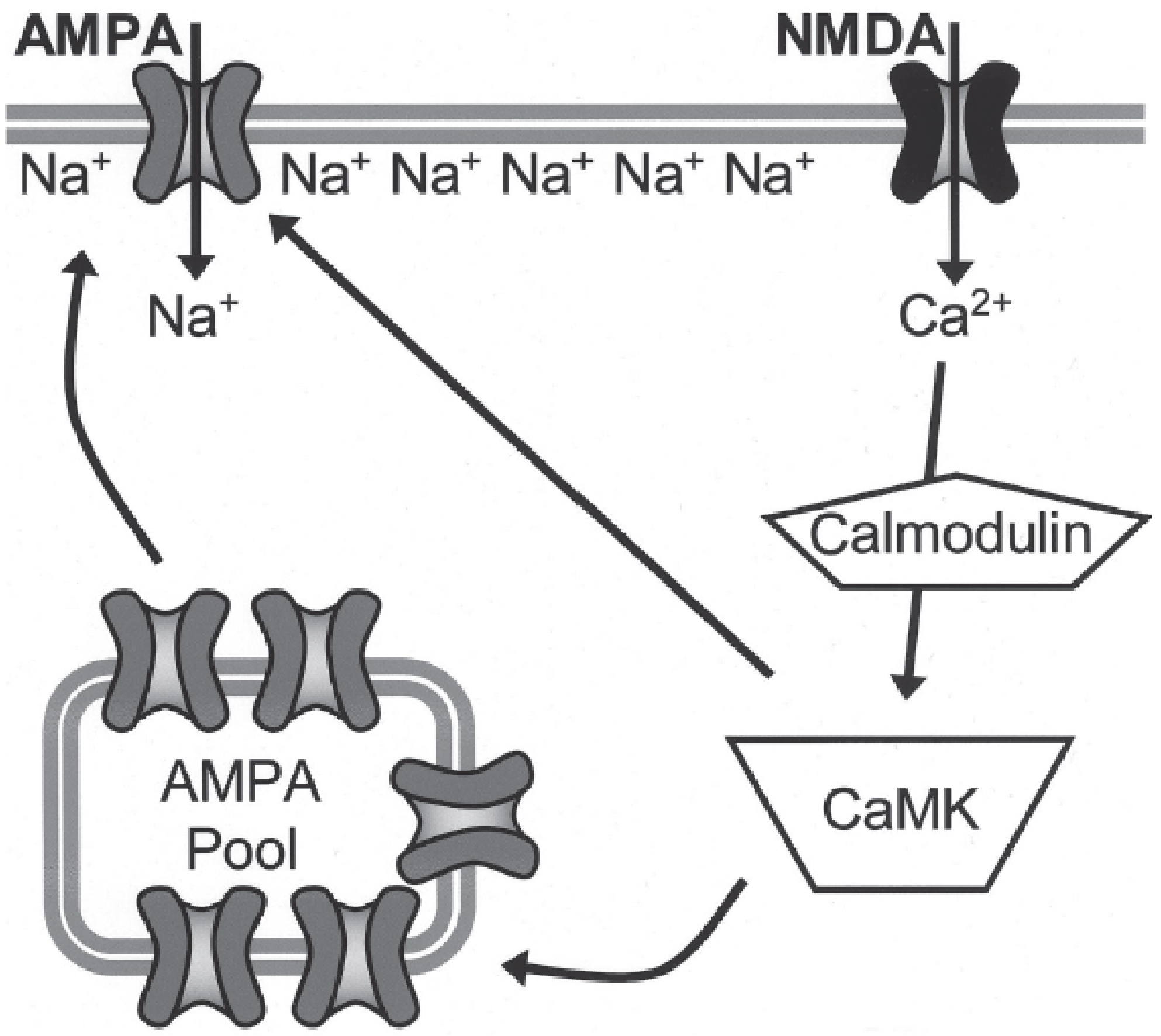
Figure: Postsynaptic mechanism of LTP. Glutamate activates AMPA receptors (Na⁺ influx, depolarization) → NMDA receptor Mg²⁺ block relieved → Ca²⁺ entry → calmodulin → CaMKII activation → AMPA receptor phosphorylation + trafficking of new AMPA receptors to the membrane. From Kaplan & Sadock's Comprehensive Textbook of Psychiatry.
Step-by-step:
- Glutamate binds to AMPA receptors → Na⁺ influx → membrane depolarization
- Sufficient depolarization expels Mg²⁺ from NMDA receptor channel
- NMDA receptor opens → large Ca²⁺ influx (the critical trigger)
- Ca²⁺ binds calmodulin → activates CaMKII (and PKC)
- CaMKII phosphorylates existing AMPA receptors → increased Na⁺ conductance
- CaMKII drives insertion of additional AMPA receptors from an intracellular pool into the postsynaptic membrane
- More AMPA receptors = larger future EPSPs = stronger synapse
Spike Timing-Dependent Plasticity (STDP)
Researchers found that the exact timing of the postsynaptic action potential matters. If a back-propagating action potential (generated in the soma, propagating back into dendrites) arrives within ~50 ms after the EPSP, NMDA receptors - which still have glutamate bound - are depolarized and open. Ca²⁺ floods in and LTP is triggered. If the spike arrives before the EPSP, LTD results instead. This is STDP - the synapse acts as a coincidence detector with millisecond-level precision.
In 2017, a distinct form called behavioral time-scale plasticity (BTSP) was discovered in hippocampal CA1, where "plateau potentials" (abrupt depolarizations with burst firing) can trigger LTP even seconds after a prior synaptic event - relevant to how place fields form during spatial navigation.
- Neuroscience: Exploring the Brain, 5th ed.
Two Phases: E-LTP and L-LTP
| Phase | Duration | Mechanism |
|---|---|---|
| Early LTP (E-LTP) | Minutes to hours | Protein synthesis-independent; CaMKII phosphorylation, AMPA receptor trafficking |
| Late LTP (L-LTP) | Days to years | Requires gene activation and new protein synthesis; structural synapse remodeling |
In L-LTP, the activated synapse receives a molecular "tag" (protein synthesis-independent). This tag allows it to capture plasticity-related proteins (PRPs) synthesized in the soma and dendrites. The tag-and-capture mechanism explains how thousands of synapses on a single neuron can be in varying states of stabilization simultaneously - the synaptic tagging hypothesis.
Key late-stage molecular players include:
- CaMKIV (nuclear) and PKA (cAMP-dependent) → phosphorylate CREB
- CREB recruits RNA polymerase II → transcription of plasticity genes (Arc, Homer, ΔFosB)
- New proteins cause dendritic spine enlargement (thin → mushroom-shaped spines) and structural synapse growth
Long-Term Depression (LTD)
LTD is the weakening of synaptic strength and is equally important as LTP - it refines neural circuits, enables forgetting of irrelevant information, and is critical for cerebellar motor learning.
The key to LTP vs. LTD is Ca²⁺ magnitude:
- High Ca²⁺ (from strong, high-frequency stimulation) → activates CaMKII → LTP
- Low Ca²⁺ (from weak, low-frequency stimulation, partial Mg²⁺ relief) → activates calcineurin (a Ca²⁺-dependent phosphatase with a higher affinity for Ca²⁺ than CaMKII) → dephosphorylates AMPA receptors → receptor endocytosis (removal from membrane) → LTD
In summary: the same NMDA receptor that drives LTP also drives LTD, with the concentration of Ca²⁺ acting as a molecular switch between kinase activation (LTP) and phosphatase activation (LTD).
- Kandel, Principles of Neural Science, 6th ed.
Homeostatic Plasticity
While Hebbian plasticity (LTP/LTD) is synapse-specific and driven by coincident activity, homeostatic plasticity operates at the whole-neuron or network level to maintain stability.
Synaptic scaling: When a neuron's overall activity is persistently too high or too low, it globally scales all its synaptic strengths up or down proportionally - preserving the relative weights while normalizing the total input. This involves adjusting the total number of AMPA receptors across all synapses.
Firing rate homeostasis: Networks regulate average firing rates through intrinsic excitability changes (ion channel expression).
Metaplasticity ("plasticity of plasticity"): Prior activity shifts the threshold for inducing future LTP or LTD. High ongoing activity raises the LTP induction threshold (making further potentiation harder), protecting against runaway excitation. This is regulated by NR2 subunit composition of NMDA receptors and other mechanisms.
"Unlike Hebbian plasticity, homeostatic plasticity is unlikely to drive information storage but rather helps maintain longer-term function in response to local changes in excitation and inhibition."
- Kaplan & Sadock's Comprehensive Textbook of Psychiatry
Clinical relevance: Ketamine (antidepressant) rapidly upregulates excitatory synaptic responses in hippocampus and cortex via non-Hebbian homeostatic mechanisms. Lithium dampens synaptic function in a similar homeostatic fashion. ECT and other brain stimulation methods are also believed to act partly through these homeostatic pathways.
Clinical and Disease Relevance
| Condition | Plasticity Abnormality |
|---|---|
| Alzheimer disease | Amyloid-β impairs NMDA receptor function and disrupts LTP |
| PTSD | Maladaptive amygdala LTP of fear memories; reconsolidation window targeted by behavioral therapy |
| Drug addiction | Psychostimulants and opiates induce LTD-like changes in nucleus accumbens during early withdrawal; LTP-like changes and mature dendritic spines after prolonged withdrawal (silent synapse mechanism) |
| Schizophrenia | NMDA receptor hypofunction impairs LTP-dependent cognition |
| Intellectual disability (e.g. fragile X) | Excessive LTD due to mGluR5 overactivation (the "mGluR theory" of fragile X) |
Summary
SYNAPTIC ACTIVITY
|
Ca²⁺ concentration in postsynaptic cell
|
HIGH Ca²⁺ LOW Ca²⁺
| |
CaMKII active Calcineurin active
| |
AMPA ↑ (LTP) AMPA ↓ (LTD)
Spine enlargement Spine shrinkage
Protein synthesis Receptor endocytosis
(if L-LTP)
Sources: Kandel, Principles of Neural Science (6th ed.); Kaplan & Sadock's Comprehensive Textbook of Psychiatry; Neuroscience: Exploring the Brain (5th ed.); Miller's Anesthesia (10th ed.); Frontiers - Hebbian and homeostatic plasticity 2026
Lipoprotic factors
Finding Sources
Finding Sources
Searching the Web
lipoprotein classes structure apolipoproteins lipid transport metabolism
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Reading File
Reading File
Loading Image
I have all the content and diagrams needed for a comprehensive answer. Let me now compose it.
Lipoprotein Structure, Classification, and Metabolism
What Are Lipoproteins?
Lipoproteins are spherical macromolecular complexes that solve a fundamental biological problem: transporting water-insoluble lipids (fats) through the aqueous bloodstream. Each particle has:
- A hydrophobic core packed with triglycerides (TG) and cholesterol esters (CE)
- A hydrophilic surface shell of phospholipids and unesterified (free) cholesterol
- Apolipoproteins embedded in the surface, which determine particle identity, dictate enzyme interactions, and serve as ligands for receptors
Classification of Lipoproteins
Lipoproteins are classified by density (measured by ultracentrifugation) - the higher the lipid-to-protein ratio, the lower the density. The major classes from largest/least dense to smallest/most dense:
| Class | Density (g/mL) | Size (nm) | Major Lipid | Key Apo | Origin | Function |
|---|---|---|---|---|---|---|
| Chylomicrons | <0.930 | 100-1000 | TG (85-90%) | ApoB-48, ApoE, ApoCII | Intestine | Transport dietary (exogenous) lipids |
| VLDL | 0.930-1.006 | 30-80 | TG (55-65%) | ApoB-100, ApoE, ApoCII | Liver | Transport hepatic (endogenous) TG to periphery |
| IDL | 1.006-1.019 | 25-35 | TG + CE (equal) | ApoB-100, ApoE | VLDL catabolism | Transitional particle; cleared by liver or converted to LDL |
| LDL | 1.019-1.063 | 18-25 | CE (~45%) | ApoB-100 only | IDL catabolism | Deliver cholesterol to peripheral tissues |
| HDL | 1.063-1.210 | 5-12 | CE + PL | ApoA-I, ApoA-II | Liver, intestine | Reverse cholesterol transport |
| Lp(a) | ~1.05 | 25-30 | CE | ApoB-100 + Apo(a) | Liver | Independent ASCVD risk factor |
Apolipoproteins: The Key Regulatory Proteins
Apolipoproteins are not merely structural - they are the functional "address labels" of lipoproteins. Most (except ApoB and Apo(a)) can transfer freely between lipoprotein particles in the blood.
| Apolipoprotein | MW (Da) | Chromosome | Carrier | Key Function |
|---|---|---|---|---|
| ApoA-I | 29,016 | 11 | HDL, chylomicrons | Core structural protein of HDL; ligand for ABCA1 (cholesterol efflux); cofactor for LCAT |
| ApoA-II | 17,414 | 1 | HDL | Structural protein of HDL (~2/3 of HDL particles) |
| ApoA-V | - | - | VLDL, chylomicrons | Promotes LPL-mediated TG lipolysis |
| ApoB-100 | 512,723 | 2 | VLDL, IDL, LDL, Lp(a) | One of the largest proteins in humans; ligand for LDL receptor; required for VLDL assembly/secretion |
| ApoB-48 | 240,800 | 2 | Chylomicrons | Intestinal form (48% of ApoB-100 sequence, from same gene via mRNA editing); required for chylomicron assembly; lacks LDL receptor-binding domain |
| ApoC-I | 6,630 | 19 | CM, VLDL, HDL | Activates LCAT; inhibits chylomicron clearance |
| ApoC-II | 8,900 | 19 | CM, VLDL, HDL | Essential cofactor for LPL (lipoprotein lipase) - without it, TG-rich lipoproteins cannot be hydrolyzed |
| ApoC-III | 8,800 | 11 | CM, VLDL, HDL | Inhibits LPL and inhibits remnant receptor binding - raises plasma TG |
| ApoE | 34,145 | 19 | CM remnants, IDL, HDL | Ligand for LDL receptor and LRP - essential for hepatic clearance of remnants |
Structural motif: Most apolipoproteins (except ApoB) contain amphipathic helices - one hydrophobic face inserts into the lipid core, the other polar face faces outward. This weak, reversible binding allows exchange between particles. ApoB, by contrast, is irreversibly embedded and cannot transfer.
- Tietz Textbook of Laboratory Medicine, 7th ed.
The Three Major Metabolic Pathways
Pathway 1: Exogenous (Dietary) Pathway - Chylomicrons
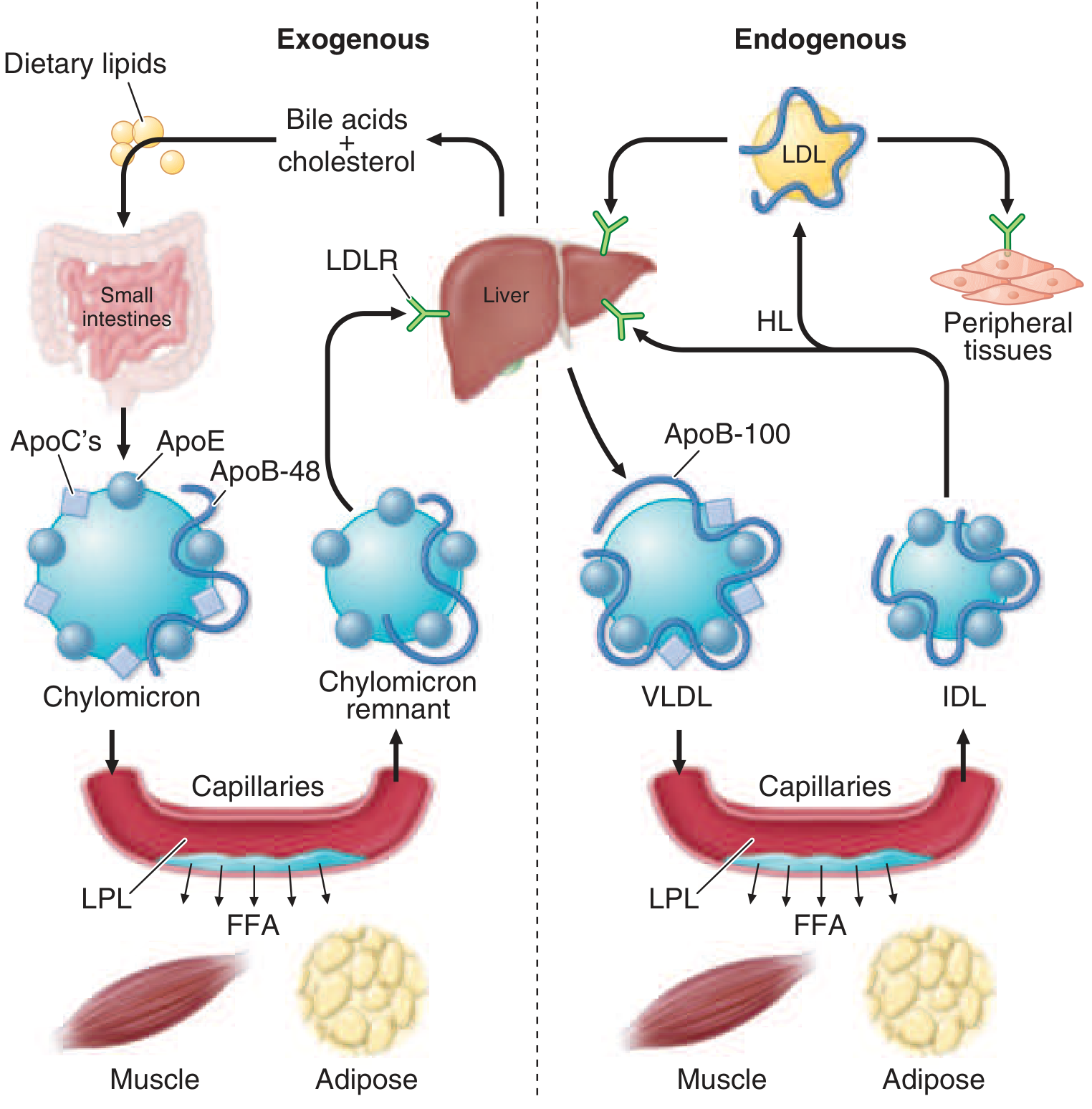
Figure: Left (exogenous): dietary lipids → chylomicrons → LPL in capillaries → FFA to muscle/adipose → chylomicron remnants → liver. Right (endogenous): liver → VLDL → LPL → IDL → LDL → peripheral tissues via LDLR. From Harrison's Principles of Internal Medicine, 22nd ed.
- Dietary fats are digested in the intestinal lumen and absorbed in the proximal small intestine
- Cholesterol and fatty acids are esterified in enterocytes; longer-chain FAs (>12C) are incorporated into TGs
- Microsomal TG transfer protein (MTP) packages TGs with ApoB-48, phospholipids, cholesteryl esters, retinyl esters, and vitamin E to form nascent chylomicrons
- Secreted into intestinal lymph → thoracic duct → systemic circulation
- In blood, chylomicrons acquire ApoC-II and ApoE from HDL
- ApoC-II activates LPL (anchored to endothelium of capillaries in adipose, heart, skeletal muscle by GPHBP1 protein) → TGs hydrolyzed → free fatty acids released to muscle (oxidation) and adipose (storage)
- Excess surface phospholipids, cholesterol, and apolipoproteins transfer to HDL
- Chylomicron remnants (enriched in cholesterol, carrying ApoB-48 + ApoE) are rapidly cleared by the liver via LRP and LDL receptor-related receptors (ApoE is the critical ligand)
- Half-life of chylomicrons < 1 hour; absent after 12-h fast
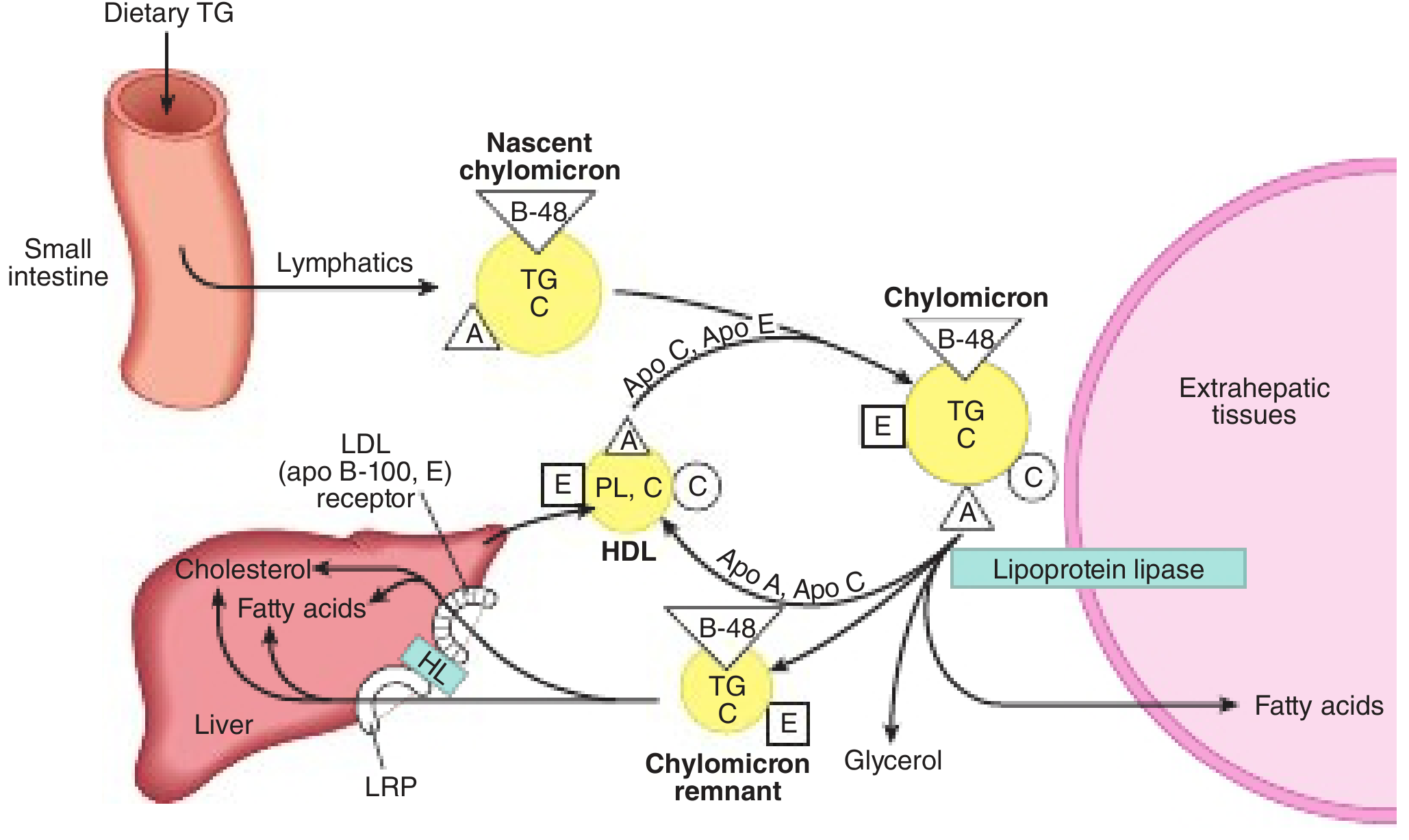
Figure: Nascent chylomicron leaves small intestine via lymphatics, acquires ApoC/E from HDL, undergoes LPL-mediated lipolysis at extrahepatic tissues, returns as chylomicron remnant to liver via LRP. From Harper's Illustrated Biochemistry, 32nd ed.
Pathway 2: Endogenous (Hepatic) Pathway - VLDL → IDL → LDL
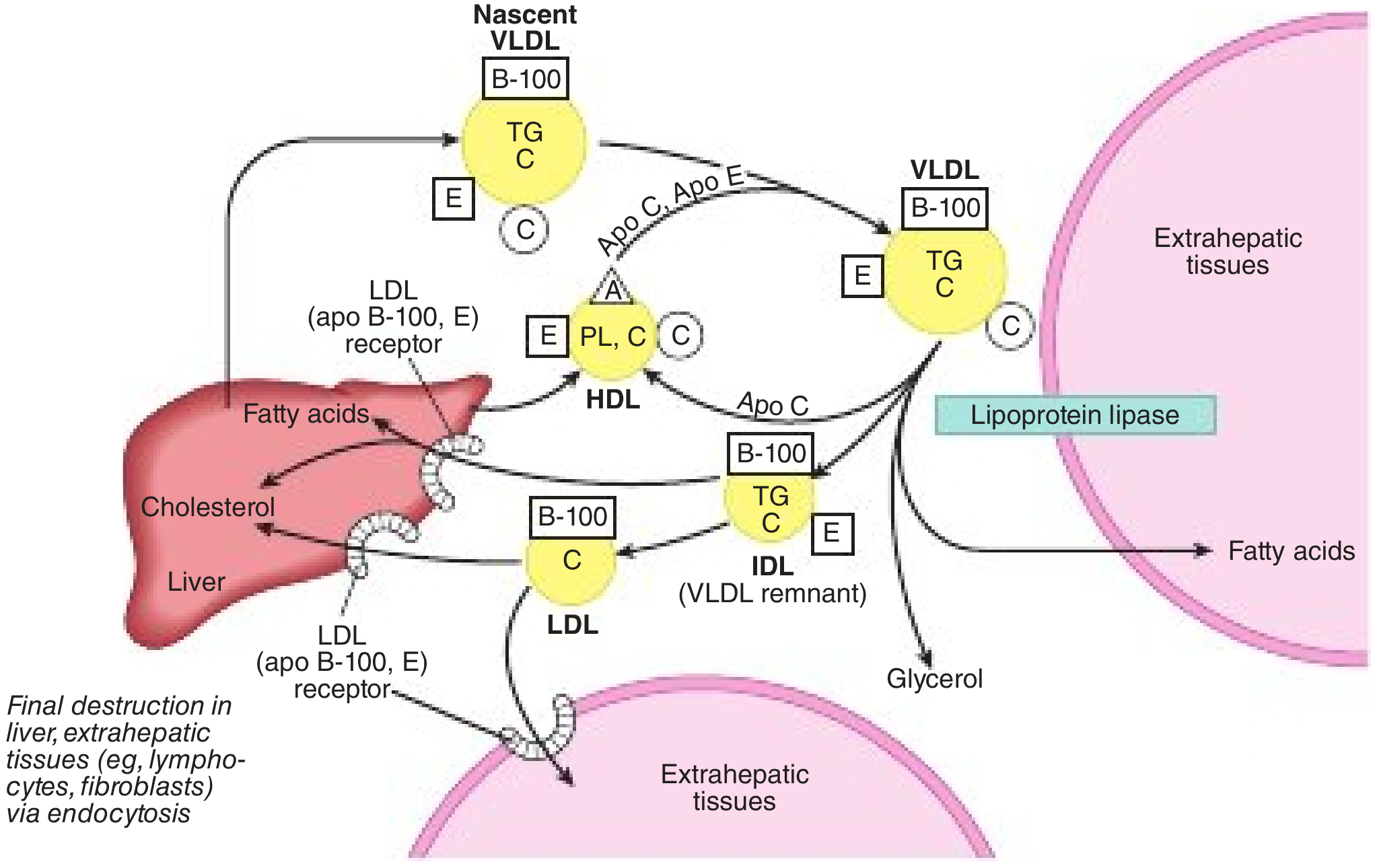
Figure: Nascent VLDL (with ApoB-100) leaves liver, acquires ApoC/E from HDL, undergoes LPL lipolysis → IDL (VLDL remnant) → either cleared by liver (via ApoE/ApoB-100 binding to LDLR) or further processed by hepatic lipase → LDL. LDL taken up in liver and extrahepatic tissues via LDLR. From Harper's Illustrated Biochemistry, 32nd ed.
- Liver assembles nascent VLDL: packages TGs (from de novo lipogenesis or fatty acids) with ApoB-100, cholesteryl esters, phospholipids via MTP → secreted into plasma
- Nascent VLDL acquires ApoC-II, ApoC-III, ApoE from circulating HDL
- LPL hydrolyzes VLDL TGs at capillary surfaces → FFAs released to tissues → VLDL shrinks to IDL (VLDL remnant)
- IDL contains ApoB-100 + ApoE → approximately half is taken up directly by the liver (via LDL receptor and LRP, using ApoE as ligand)
- Remaining IDL is further hydrolyzed by hepatic lipase (HL) → most apolipoproteins (including ApoE) transferred to HDL, leaving only ApoB-100 → LDL is formed
- LDL circulates with a half-life of ~2-3 days, delivering cholesterol to:
- Liver (~70%) via LDL receptor (LDLR) - ApoB-100 is the ligand
- Peripheral tissues (lymphocytes, fibroblasts, adrenal glands, etc.) via endocytosis
LDL Receptor (LDLR) Regulation (Brown & Goldstein pathway):
- When cell cholesterol is adequate → LDLR synthesis is suppressed (SREBP pathway inactive)
- When cell cholesterol is low → LDLR expression increases → more LDL uptake
- This is the target of statins (inhibit HMG-CoA reductase → lower intracellular cholesterol → upregulate LDLR → lower plasma LDL)
- Familial hypercholesterolemia = loss-of-function mutations in LDLR → markedly elevated LDL
Pathway 3: Reverse Cholesterol Transport - HDL
HDL is responsible for transporting excess cholesterol from peripheral tissues (including arterial wall macrophages) back to the liver for excretion - the anti-atherogenic "reverse cholesterol transport."
- Nascent HDL (disc-shaped, lipid-poor) is secreted by the liver and intestine, carrying ApoA-I
- ABCA1 transporter on macrophages and other peripheral cells effluxes free cholesterol and phospholipids onto ApoA-I → forming nascent HDL discs (defective in Tangier disease)
- LCAT (lecithin-cholesterol acyltransferase), activated by ApoA-I, esterifies free cholesterol → cholesteryl esters sink into the core → HDL matures into spherical HDL3 → HDL2
- CETP (cholesterol ester transfer protein) transfers CE from HDL to VLDL/chylomicrons in exchange for TGs (allowing indirect return of cholesterol to liver)
- SR-BI (scavenger receptor class B) on hepatocytes takes up CE selectively from HDL without degrading the particle ("selective uptake")
- Phospholipid transfer protein (PLTP) transfers phospholipids between lipoproteins; hepatic lipase and endothelial lipase (EL) remodel HDL, generating smaller particles
Lipoprotein(a) - Lp(a)
Lp(a) is a special atherogenic lipoprotein:
-
Structure: LDL-like particle with ApoB-100 covalently linked (via single disulfide bond) to Apo(a) - a large protein with kringle domains homologous to plasminogen
-
Synthesized in the liver; plasma levels are >90% genetically determined (by LPA gene kringle copy number)
-
Pathogenic mechanisms:
- Atherogenic (like LDL, deposits cholesterol in arterial wall)
- Thrombogenic: Apo(a) competitively inhibits fibrinolysis by interfering with plasminogen activation
-
Levels >125 nmol/L (or >50 mg/dL) are an independent ASCVD and aortic stenosis risk factor
-
Not reduced by statins (may modestly increase); niacin reduces it ~20-30%; PCSK9 inhibitors reduce ~25%; antisense oligonucleotides (pelacarsen) and small interfering RNA (olpasiran) reduce Lp(a) by >80% - now in phase 3 trials
-
Harrison's Principles of Internal Medicine, 22nd ed.; Lp(a) in clinical practice - CCJM 2026
Friedewald Formula and Clinical Measurement
From Harrison's:
LDL-C = Total Cholesterol - (TG/5) - HDL-C
The TG/5 term estimates VLDL-C (based on the 5:1 TG-to-cholesterol ratio in VLDL). This formula is invalid when TG > 400 mg/dL (e.g. in severe hypertriglyceridemia or type III hyperlipoproteinemia).
Non-HDL-C = Total Cholesterol - HDL-C (captures all atherogenic apoB-containing particles: VLDL + IDL + LDL + Lp(a)).
There is growing evidence that ApoB measurement directly (one molecule per atherogenic particle) may better predict ASCVD risk than LDL-C alone.
Summary: The Flow of Lipid Traffic
DIETARY LIPIDS HEPATIC LIPIDS
| |
Chylomicrons Nascent VLDL
(ApoB-48) (ApoB-100)
| |
LPL (ApoC-II) LPL (ApoC-II)
| |
Chylomicron IDL (VLDL remnant)
remnants → Liver Liver (50%) + HL
(ApoE → LRP) |
LDL
(ApoB-100 → LDLR)
|
Peripheral tissues
|
HDL (reverse transport)
ApoA-I → ABCA1 → LCAT
|
Liver (SR-BI, CETP)
Sources: Harrison's Principles of Internal Medicine (22nd ed.); Harper's Illustrated Biochemistry (32nd ed.); Tietz Textbook of Laboratory Medicine (7th ed.); Henry's Clinical Diagnosis and Management by Laboratory Methods
This is a shared conversation. Sign in to Orris to start your own chat.