Emphysema from rhobbin
Reading File
Finding Sources
Searching the Web
emphysema "Robin sequence" OR "Pierre Robin" OR "rho kinase" OR "robbing" mechanism
Finding Sources
Finding Sources
Finding Sources
Searching the Web
emphysema "rhobbin" OR "robbin" pathology OR pulmonology
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
I have everything I need for a comprehensive answer. Let me now compose the full response from Robbins & Cotran Pathologic Basis of Disease and Robbins & Kumar Basic Pathology.
Emphysema (Robbins Pathology)
"Rhobbin" = Robbins - the classic pathology textbook series (Robbins & Cotran / Robbins & Kumar Basic Pathology).
Definition
Emphysema is defined as irreversible enlargement of the airspaces distal to the terminal bronchiole, accompanied by destruction of alveolar walls, without significant fibrosis. The key distinction from other lung diseases is the anatomic definition - it is classified by where in the acinus the destruction occurs.
- Robbins & Cotran Pathologic Basis of Disease, p. 634
- Robbins & Kumar Basic Pathology, p. 448
Types of Emphysema
The classification is based on the part of the acinus (the respiratory unit distal to the terminal bronchiole) that is destroyed.
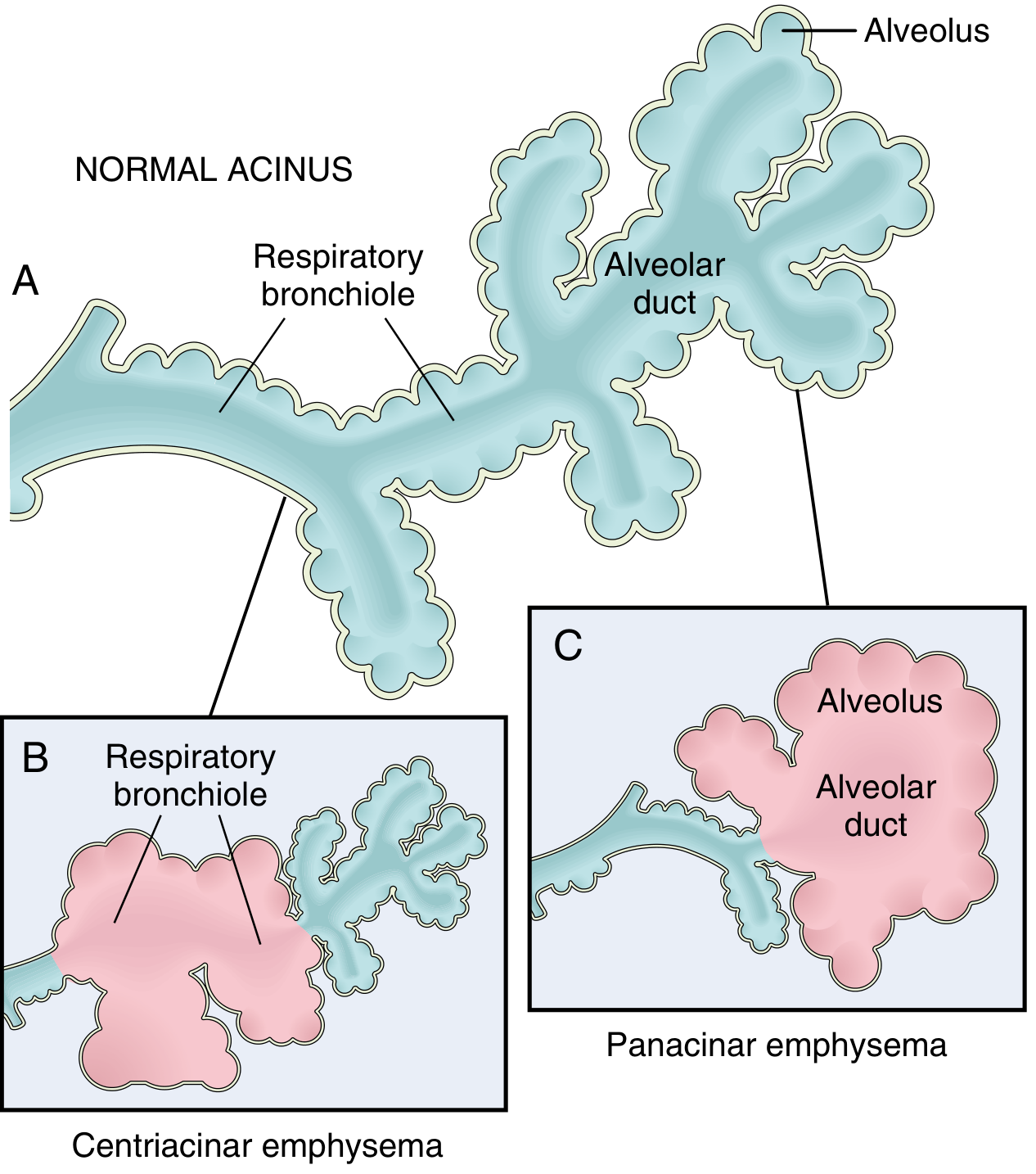
Fig. 15.6 from Robbins & Cotran: Normal acinus (A), Centriacinar emphysema (B), Panacinar emphysema (C)
1. Centriacinar (Centrilobular) Emphysema
- Most common type - >95% of clinically significant cases
- The central/proximal parts of the acinus (respiratory bronchioles) are destroyed; distal alveoli are spared
- Both emphysematous and normal airspaces coexist within the same acinus/lobule
- Upper lobe predominant, especially apical segments
- Strongly associated with cigarette smoking
2. Panacinar (Panlobular) Emphysema
- The entire acinus is uniformly enlarged - from respiratory bronchiole to terminal alveoli
- Lower lobe predominant, especially at the bases
- Associated with α1-antitrypsin (α1-AT) deficiency and exacerbated by smoking
- About 1% of emphysema patients have α1-AT deficiency; >80% of homozygous Z-allele individuals develop panacinar emphysema
3. Distal Acinar (Paraseptal) Emphysema
- The distal portion of the acinus is affected; proximal portion is spared
- Occurs near pleura, along lobular connective tissue septa, adjacent to fibrosis/scarring
- More severe in upper half of lungs
- Enlarged airspaces range from <0.5 cm to >2.0 cm - can form cystic structures/bullae
- Classic cause of spontaneous pneumothorax in young adults
4. Irregular (Paracicatricial) Emphysema
- The acinus is irregularly involved
- Almost always associated with scarring
- Usually occurs in small foci and is clinically insignificant
Only centriacinar and panacinar types cause clinically significant airflow obstruction and COPD.
Pathogenesis
The two main drivers are cigarette smoking and α1-antitrypsin deficiency, both converging on alveolar wall destruction.
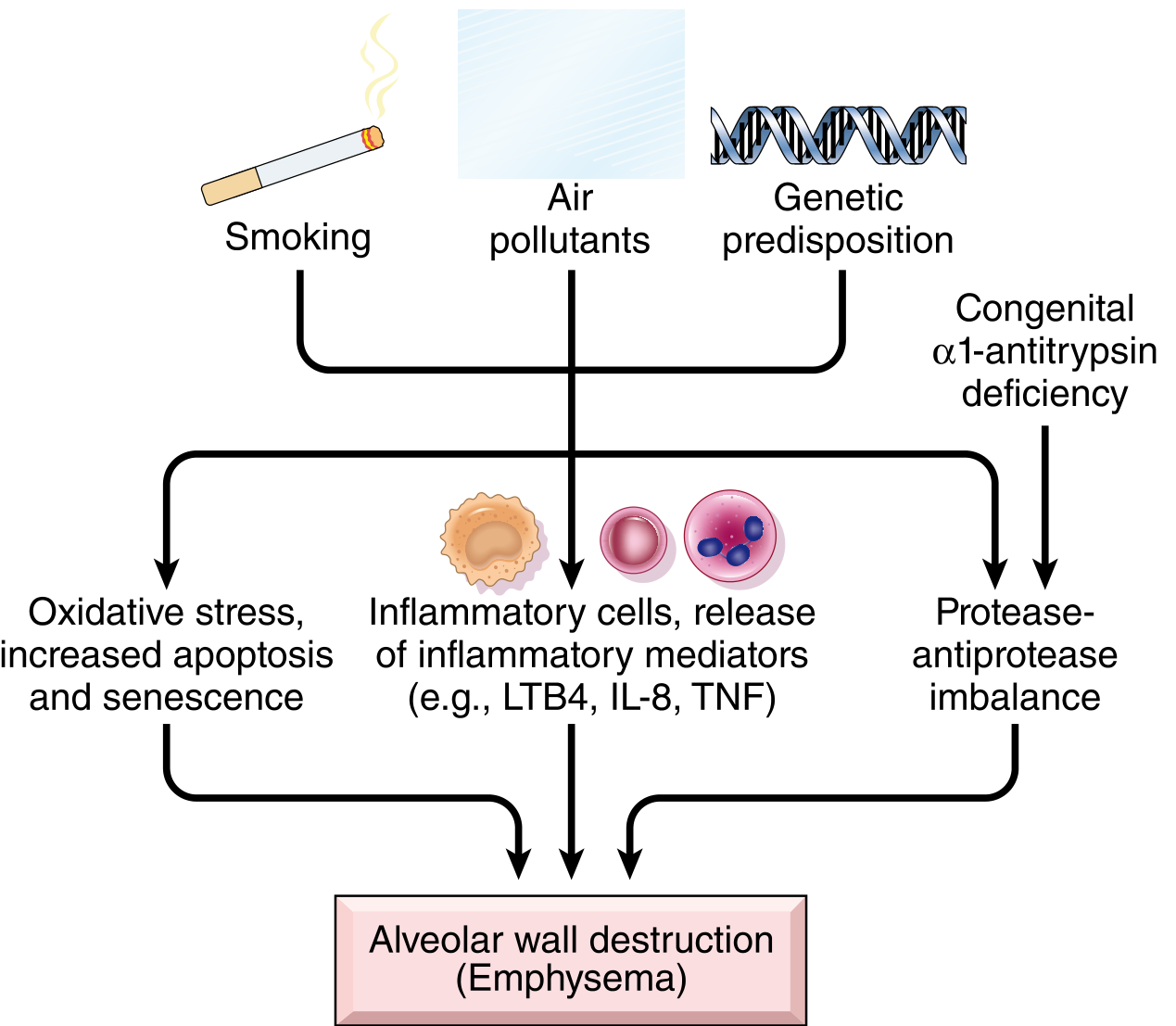
Fig. 15.8 from Robbins & Cotran: Pathogenesis of emphysema
Mechanisms (Robbins & Cotran, p. 635-636):
1. Toxic injury and inflammation
- Cigarette smoke and noxious particles damage respiratory epithelium and trigger chronic inflammation
- Inflammatory mediators released: LTB4, IL-8, TNF, and others
- Macrophages and resident epithelial cells recruit more neutrophils and amplify inflammation
- T and B cells accumulate in affected lung (role of adaptive immunity still uncertain)
2. Protease-antiprotease imbalance (the central mechanism)
- Inflammatory cells release proteases (especially elastase) that break down connective tissue (elastin, collagen)
- Normally, α1-antitrypsin inhibits these proteases - but in smokers or those with genetic deficiency, this protection is overwhelmed
- Loss of elastic tissue in alveolar walls reduces radial traction on small airways → respiratory bronchioles collapse during expiration → functional airflow obstruction without mechanical obstruction
- α1-AT is encoded at the Pi locus on chromosome 14; ~0.01% of the U.S. population is homozygous for the Z allele (very low serum α1-AT)
3. Oxidative stress
- Tobacco smoke and inflammatory cells produce oxidants causing tissue damage, endothelial dysfunction, and further inflammation
- NRF2 (encoded by NFE2L2) is a key transcription factor protecting against oxidant damage; genetic variants in NRF2 pathways are linked to smoking-related lung disease
4. Infection
- Bacterial/viral infections are not thought to initiate emphysema but can acutely exacerbate existing disease
Morphology
Gross:
- Advanced emphysema → voluminous, pale lungs that overlap the heart and flatten the diaphragm (especially panacinar)
- Centriacinar emphysema: lungs deeper pink, less voluminous; upper 2/3 more affected
- Large alveoli visible on cut surface of fixed lungs
- Apical blebs or bullae may appear (especially paraseptal/irregular emphysema)
Microscopic:
- Destruction of alveolar walls without fibrosis → enlarged airspaces
- Focal centriacinar fibrosis may be seen
- Loss of attachments between alveoli and small airway walls
- Pores of Kohn become so large that septa appear to "float" or protrude blindly into airspaces with a club-shaped end
- Decreased capillary bed area (leads to pulmonary hypertension in severe disease)
- Bronchiolar inflammation and submucosal fibrosis in advanced disease
Key Summary Table
| Feature | Centriacinar | Panacinar | Distal Acinar | Irregular |
|---|---|---|---|---|
| Location in acinus | Central (respiratory bronchiole) | Entire acinus | Distal part | Irregular |
| Lung zone | Upper lobes | Lower lobes/bases | Near pleura, upper | Near scars |
| Main cause | Smoking | α1-AT deficiency | Unknown | Scarring |
| COPD significance | Yes (most common) | Yes | No | No |
| Classic presentation | Heavy smoker | Young/non-smoker | Spontaneous pneumothorax | Incidental |
Sources: Robbins & Cotran Pathologic Basis of Disease, pp. 634-636 | Robbins & Kumar Basic Pathology, pp. 448-449
Chronic bronchitis
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Loading Image
I have everything needed for a complete Robbins-based answer on chronic bronchitis.
Chronic Bronchitis (Robbins Pathology)
Definition
Chronic bronchitis is defined clinically (not anatomically) as:
Persistent productive cough for at least 3 consecutive months in at least 2 consecutive years, in the absence of any other identifiable cause.
This clinical definition contrasts with emphysema, which is defined anatomically. Long-standing disease can lead to progressive lung dysfunction, hypoxemia, pulmonary hypertension, and cor pulmonale.
- Robbins & Cotran Pathologic Basis of Disease, p. 637
- Robbins & Kumar Basic Pathology, p. 448
Etiology
- Cigarette smoking is the primary cause - 90% of affected individuals are smokers
- Other inhaled irritants: sulfur dioxide, nitrogen dioxide, grain dust, cotton dust, silica
- Cigarette smoke damages airways in two key ways:
- Causes direct cellular damage and chronic inflammation
- Impairs ciliary action of respiratory epithelium, preventing mucus clearance and increasing infection risk
Pathogenesis
1. Mucus Hypersecretion (the earliest and most distinctive feature)
The initiating event is hypersecretion of mucus in the large airways, driven by:
- Enlargement of submucosal glands in the trachea and bronchi (measured by the Reid index)
- Goblet cell hyperplasia in small bronchi and bronchioles - causes mucus production that obstructs small airways
- Mediators involved: histamine and IL-13 from T cells
- Both gland enlargement and goblet cell increase are considered protective reactions against tobacco smoke/pollutants
2. Acquired CFTR Dysfunction
- Smoking leads to acquired CFTR (cystic fibrosis transmembrane conductance regulator) dysfunction
- This causes secretion of abnormal, dehydrated mucus that worsens disease severity
3. Inflammation
- Inhaled irritants cause cellular damage → both acute and chronic inflammatory responses
- Inflammatory cells: neutrophils, lymphocytes, macrophages (notably, NO eosinophils - unlike asthma)
- Long-standing inflammation causes small airway fibrosis (bronchi and bronchioles <2-3 mm diameter) → chronic airway obstruction
4. Infection
- Does not initiate chronic bronchitis
- Plays a role in maintaining the disease and triggering acute exacerbations
Morphology
Gross
- Mucosal lining of larger airways: hyperemic, swollen, edematous, covered by mucinous or mucopurulent secretions
- Smaller bronchi and bronchioles filled with secretions
- Heavy casts of secretions and pus can fill bronchi/bronchioles in severe cases
Microscopic
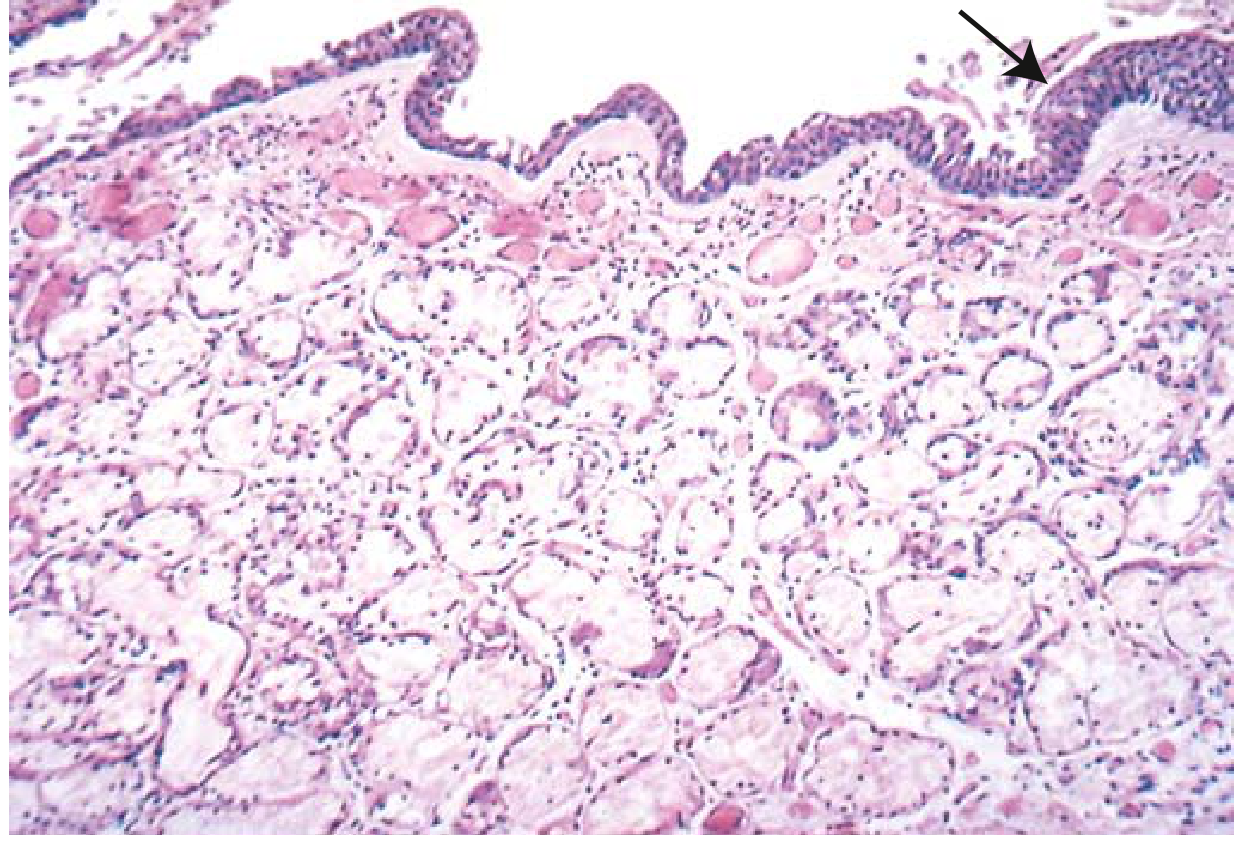
FIG. 11.8 from Robbins & Kumar: Chronic bronchitis. Note the markedly thickened mucous gland layer (approximately twice normal) and squamous metaplasia (arrow).
Key microscopic findings:
- Chronic inflammation of airways (lymphocytes and macrophages predominate)
- Enlargement of mucus-secreting submucosal glands - the hallmark diagnostic feature
- Goblet cell hyperplasia in small airways
- Thickening of bronchiolar wall - smooth muscle hypertrophy, ECM deposition, peribronchial fibrosis
- Mucus plugging of bronchiolar lumens
- In severe cases: bronchiolitis obliterans - complete lumen obliteration by fibrosis
- Emphysematous changes frequently coexist
The Reid Index
Reid index = thickness of submucosal gland layer / thickness of bronchial wall (epithelium to cartilage)
| Value | |
|---|---|
| Normal | ≤ 0.4 |
| Chronic bronchitis | > 0.4 (increased in proportion to severity and duration) |
Clinical Features
- Most patients have a smoking history of ≥40 pack-years
- Presents insidiously with:
- Chronic cough with sputum production (cardinal symptom)
- Increasing dyspnea on exertion
- Wheezing/bronchospasm in some (asthmatic bronchitis)
- Symptoms are often worse in the morning
- Key diagnostic test: spirometry showing FEV1/FVC < 0.7
"Blue Bloater" vs "Pink Puffer"
Robbins describes the classic clinical contrast between the two ends of the COPD spectrum:
| Feature | "Blue Bloater" (Chronic Bronchitis) | "Pink Puffer" (Emphysema) |
|---|---|---|
| Age at presentation | 40-45 years | 50-75 years |
| Dominant symptom | Cough, copious sputum | Dyspnea (early, severe) |
| Cyanosis | Yes (hypoxic) | No (well-oxygenated at rest) |
| Dyspnea | Mild, late | Severe, early |
| Infections | Common | Occasional |
| Cor pulmonale | Common | Uncommon (end-stage only) |
| Airway resistance | Increased | Normal or slightly increased |
| Elastic recoil | Normal | Low |
| Chest X-ray | Prominent vessels; large heart | Hyperinflation; normal heart size |
Most real patients fall somewhere in the middle, with a mix of bronchitic and emphysematous changes.
Complications and Prognosis
- Pulmonary hypertension due to chronic hypoxemia-induced vasoconstriction + loss of capillary beds
- Cor pulmonale (right heart failure) - more common in bronchitis-predominant COPD
- Acute respiratory failure from superimposed infections
- Obstructive sleep apnea - ~10-30% of patients
- Patients with severe chronic bronchitis have more frequent exacerbations, faster progression, and worse outcomes than those with pure emphysema
Treatment options: Smoking cessation, oxygen therapy, long-acting bronchodilators with inhaled corticosteroids, antibiotics, physical therapy, and in selected patients: lung volume reduction surgery or lung transplantation.
Sources: Robbins & Cotran Pathologic Basis of Disease, pp. 637-638 | Robbins & Kumar Basic Pathology, pp. 448-450
Chronic bronchitis
The previous answer already covered chronic bronchitis from Robbins in full detail. Here's a concise summary in case you need a quick reference:
Chronic Bronchitis - Quick Reference (Robbins)
Definition
Persistent productive cough for ≥3 months in ≥2 consecutive years, with no other identifiable cause. Defined clinically (not anatomically).
Etiology
- Cigarette smoking (#1 cause - 90% of cases)
- Air pollutants: SO₂, NO₂, grain/cotton/silica dust
Pathogenesis
| Mechanism | Details |
|---|---|
| Mucus hypersecretion | Submucosal gland hypertrophy + goblet cell hyperplasia; driven by histamine, IL-13 |
| Acquired CFTR dysfunction | Smoking → dehydrated, abnormal mucus |
| Inflammation | Neutrophils, lymphocytes, macrophages (no eosinophils) → small airway fibrosis |
| Infection | Maintains disease and triggers exacerbations; does NOT initiate it |
Reid Index
Submucosal gland thickness / Bronchial wall thickness
- Normal: ≤ 0.4
- Chronic bronchitis: > 0.4
Morphology
- Gross: Hyperemic, edematous mucosa; mucopurulent secretions; mucus plugs
- Micro: Submucosal gland enlargement, goblet cell hyperplasia, chronic inflammation (lymphocytes/macrophages), bronchiolar wall fibrosis
- Severe cases: bronchiolitis obliterans (lumen obliteration by fibrosis)
Clinical
- Cough with sputum production (worse in morning)
- FEV1/FVC < 0.7 on spirometry
- Classic picture: "Blue Bloater" - cyanotic, hypercapnic, overweight, recurrent infections, cor pulmonale
Complications
Pulmonary hypertension → cor pulmonale → right heart failure; acute respiratory failure from superimposed infections
If you wanted this covered from a different angle (clinical management, comparison with asthma, etc.), just let me know.
Asthma
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
I have comprehensive content from both Robbins books. Now composing the full answer.
Asthma (Robbins Pathology)
Definition
Asthma is a chronic inflammatory disorder of the airways that causes recurrent, episodic bronchospasm. It is characterized by:
- Intermittent, reversible airway obstruction
- Chronic bronchial inflammation with eosinophils
- Bronchial smooth muscle hypertrophy and hyperreactivity
- Increased mucus secretion
Symptoms include wheezing, breathlessness, chest tightness, and cough - typically worse at night or early morning. Between attacks, patients may be virtually asymptomatic.
- Robbins & Cotran Pathologic Basis of Disease, p. 638
- Robbins & Kumar Basic Pathology, p. 451
Types of Asthma
1. Atopic Asthma (most common)
- Classic type I IgE-mediated hypersensitivity reaction
- Begins in childhood; positive family history of atopy
- Associated with: allergic rhinitis, urticaria, eczema
- Triggers: dust, pollen, animal dander, food, viral infections
- Skin test with offending allergen → immediate wheal-and-flare reaction
- Diagnosis: elevated total serum IgE; RAST (radioallergosorbent test) identifies specific IgE antibodies
2. Nonatopic Asthma
- No evidence of allergen sensitization; skin tests usually negative
- Less frequent family history
- Triggers: viral respiratory infections (rhinovirus, parainfluenza, RSV), air pollutants (tobacco smoke, SO₂, ozone, NO₂), cold air, exercise
3. Drug-Induced Asthma
- Aspirin-sensitive asthma: associated with recurrent rhinitis and nasal polyps
- Aspirin/NSAIDs inhibit COX → ↓ prostaglandin E₂ → ↑ leukotrienes (LTB4, LTC4, LTD4, LTE4) → bronchoconstriction
- Triggers urticaria AND asthmatic attacks
4. Occupational Asthma
- Triggered by workplace fumes, organic/chemical dusts, gases, or chemicals (epoxy resins, wood dust, toluene, formaldehyde, penicillin products)
- Only minute quantities needed; attacks occur after repeated exposure
- Mechanisms: type I reactions, direct bronchoconstriction, or unknown hypersensitivity
Pathogenesis
The core abnormality in atopic asthma is an exaggerated Th2 immune response to normally harmless environmental antigens.
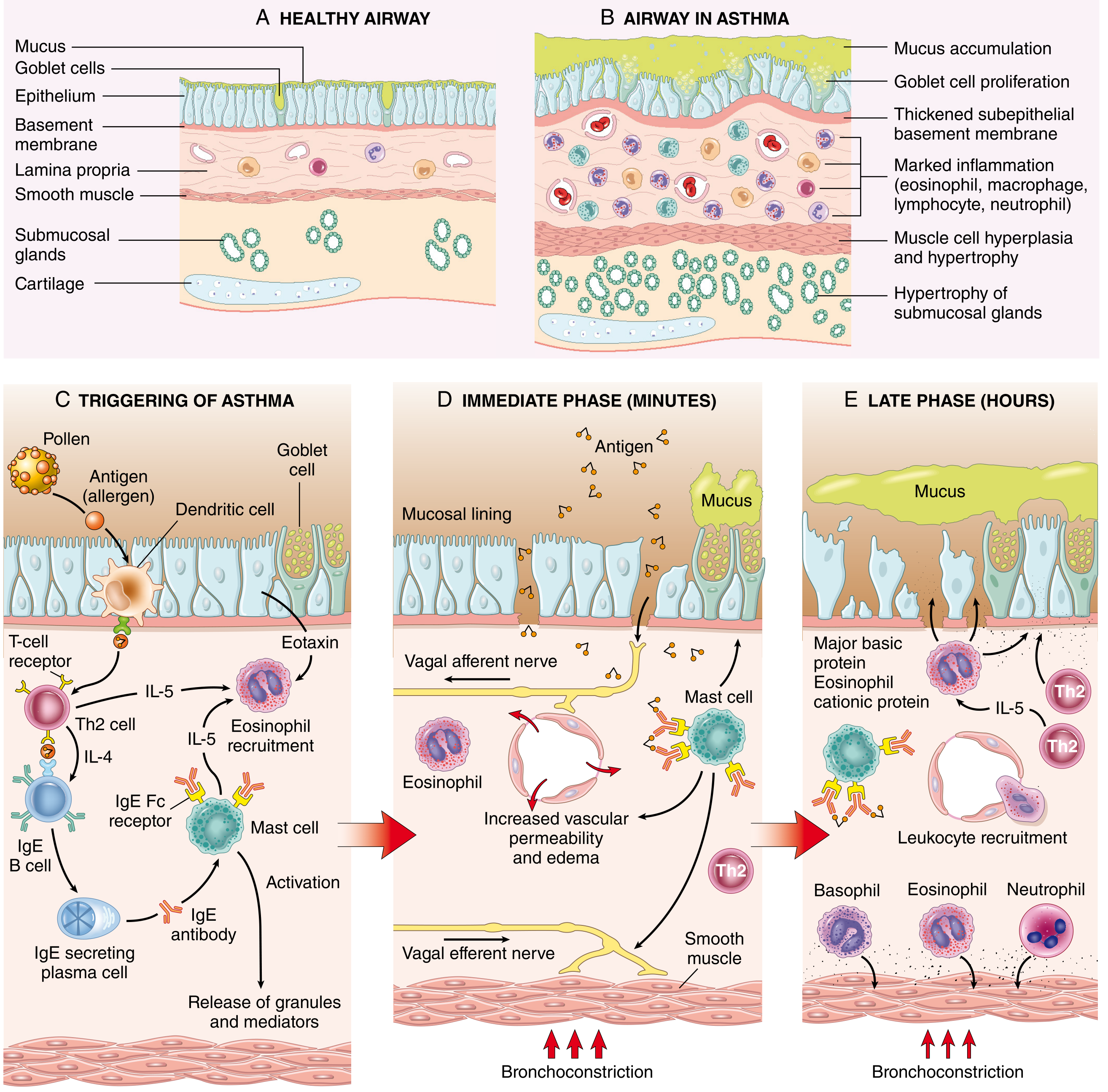
Fig. 15.10 from Robbins & Cotran: (A-B) Normal vs. asthmatic airway; (C) Allergen triggering Th2/IgE response; (D) Immediate phase - mast cell degranulation → bronchoconstriction; (E) Late phase - leukocyte recruitment → sustained inflammation
Step-by-Step Mechanism
1. Sensitization
- Inhaled allergens are processed by dendritic cells → presented to naive T cells → differentiation into Th2 cells
- Th2 cytokines drive the response:
- IL-4 → stimulates B cells to produce IgE
- IL-5 → recruits and activates eosinophils
- IL-13 → stimulates mucus secretion from submucosal glands; promotes IgE production
- IgE binds to Fc receptors on submucosal mast cells (sensitization complete)
- ILC2 (group 2 innate lymphoid cells) also produce Th2-type cytokines, activated by damaged epithelial cytokines (IL-33, TSLP) - this is called type 2 immunity
2. Early Phase Reaction (minutes)
- Re-exposure to allergen → cross-linking of IgE on mast cells → degranulation
- Mediators released:
- Histamine - bronchoconstriction
- Prostaglandin D₂ - bronchoconstriction + vasodilation
- Leukotrienes C4, D4, E4 - prolonged bronchoconstriction, ↑ vascular permeability, ↑ mucus secretion
- Also activated: vagal (parasympathetic) afferent/efferent reflexes → smooth muscle contraction via muscarinic receptors
- Net result: bronchoconstriction, mucus production, vasodilation, edema
3. Late Phase Reaction (hours)
- Inflammatory mediators from mast cells + epithelial cells release chemokines (including eotaxin)
- Recruitment of: eosinophils, neutrophils, Th2 cells, basophils, monocytes
- Eosinophils release:
- Major basic protein (MBP)
- Eosinophil cationic protein (ECP) - both damage the epithelium
- Galectin-10 (GAL10) → forms Charcot-Leyden crystals → pro-inflammatory, induces mucus production
- IL-5 antagonists are effective in severe eosinophilic asthma → confirms IL-5's central role
4. Airway Remodeling (chronic, repeated bouts)
- Hypertrophy and hyperplasia of bronchial smooth muscle
- Subepithelial fibrosis (type I and III collagen deposition)
- Increased vascularity
- Mucous gland enlargement + goblet cell hyperplasia
- Thickened subepithelial basement membrane
- May contribute to chronic, irreversible airway obstruction over time
Genetics
Key genetic associations:
- Chromosome 5q locus near cytokine cluster: IL-3, IL-4, IL-5, IL-9, IL-13 and IL-4 receptor
- IL-13 gene polymorphisms: strongest, most consistent association
- IL-4 receptor variants: linked to atopy, elevated IgE, asthma
- HLA class II alleles - linked to IgE production against specific antigens (e.g., ragweed pollen)
- IL-33 / ST2 receptor variants - induce Th2 cytokine production
- TSLP (thymic stromal lymphopoietin) gene variants - epithelial cytokine that initiates allergic reactions
Hygiene Hypothesis
The rising incidence of asthma in affluent/urban societies is partly explained by the hygiene hypothesis: lack of microbial exposure in early childhood leads to failure to develop immune tolerance → hyperreactivity to allergens later in life. Still no clear mechanistic proof, but has spurred trials of probiotics and early allergen exposure.
Morphology
Gross (status asthmaticus / fatal cases)
- Lungs overinflated, small areas of atelectasis
- Bronchi and bronchioles occluded by thick, tenacious mucus plugs (often contain shed epithelium)
Microscopic - Airway Remodeling
| Finding | Details |
|---|---|
| Mucus plugs | Contain shed epithelium, eosinophils |
| Curschmann spirals | Twisted mucus plugs from subepithelial gland ducts |
| Charcot-Leyden crystals | Bipyramidal crystals of galectin-10 from eosinophils |
| Eosinophilia | Prominent in sputum and tissue |
| Thickened airway wall | Due to smooth muscle hypertrophy + edema |
| Sub-basement membrane fibrosis | Type I and III collagen; hallmark of remodeling |
| Increased vascularity | Angiogenesis |
| Goblet cell hyperplasia | ↑ mucus production |
| Smooth muscle hypertrophy/hyperplasia | ↑ bronchospasm tendency |
Key Mediators Summary
| Mediator | Source | Role | Pharmacologic significance |
|---|---|---|---|
| Leukotrienes C4/D4/E4 | Mast cells, eosinophils | Prolonged bronchoconstriction, ↑ permeability, ↑ mucus | Leukotriene antagonists effective |
| Acetylcholine | Parasympathetic nerves | Smooth muscle constriction via muscarinic receptors | Anticholinergics effective |
| IL-5 | Th2 cells, ILC2 | Eosinophil recruitment and activation | Anti-IL-5 biologics (mepolizumab) effective |
| Galectin-10 / Charcot-Leyden crystals | Eosinophils | Pro-inflammatory; ↑ mucus production | Under investigation |
| Histamine | Mast cells | Bronchoconstriction | Minor role - antihistamines not very effective |
| Prostaglandin D₂ | Mast cells | Bronchoconstriction + vasodilation | Minor role |
| IL-4, IL-13 | Th2 cells | IgE production, mucus secretion | Anti-IL-4R biologics (dupilumab) effective |
Clinical Features
- Classic acute attack: chest tightness, dyspnea, wheezing, cough - lasts up to several hours
- Prolonged expiration (wheeze on expiration)
- Diagnosis: spirometry showing reversible airflow obstruction; peripheral eosinophilia in atopic asthma; Curschmann spirals and Charcot-Leyden crystals in sputum
- Acute severe asthma (status asthmaticus): unrelenting attack lasting days or weeks; can be fatal with marked cyanosis
Treatment
- Bronchodilators (β₂-agonists, anticholinergics)
- Glucocorticoids (inhaled/systemic)
- Leukotriene antagonists (montelukast)
- Biologics for severe/refractory disease:
- Anti-IL-4R (dupilumab)
- Anti-IL-5 (mepolizumab, reslizumab)
- Anti-TSLP (tezepelumab)
- ~50% of childhood asthma remits in adolescence; may return in adulthood
Sources: Robbins & Cotran Pathologic Basis of Disease, pp. 638-643 | Robbins & Kumar Basic Pathology, pp. 451-455
Bronchiectisis
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
All the content I need is in hand. Here is the full answer.
Bronchiectasis (Robbins Pathology)
Definition
Bronchiectasis is the permanent dilation of bronchi and bronchioles caused by destruction of smooth muscle and supporting elastic tissue by inflammation, resulting from persistent or severe infections. It is not a primary disorder - it always occurs secondary to persistent infection or obstruction.
Cardinal symptoms: chronic cough + expectoration of copious purulent (often foul-smelling) sputum.
- Robbins & Cotran Pathologic Basis of Disease, p. 642
- Robbins & Kumar Basic Pathology, p. 455
Conditions That Predispose to Bronchiectasis
1. Congenital / Hereditary Conditions
| Condition | Mechanism |
|---|---|
| Cystic fibrosis | Abnormally viscid mucus → obstruction → recurrent infections → widespread severe bronchiectasis |
| Primary ciliary dyskinesia (immobile cilia syndrome) | Autosomal recessive; mutations in ciliary motor proteins (e.g., dynein) → impaired mucociliary clearance → recurrent infections |
| Kartagener syndrome | ~50% of primary ciliary dyskinesia cases; situs inversus + bronchiectasis + sinusitis; males infertile due to sperm dysmotility |
| Immunodeficiency states | Particularly immunoglobulin deficiencies → recurrent bacterial infections → localized or diffuse bronchiectasis |
| Intralobar lung sequestration | Chronic infection in non-communicating lung segment |
2. Bronchial Obstruction
- Causes: tumors, foreign bodies, mucus impaction
- Result: localized bronchiectasis in the obstructed segment
- May also complicate atopic asthma and chronic bronchitis
3. Necrotizing / Suppurative Pneumonia
- Single severe episode or recurrent infections
- Virulent organisms: Staphylococcus aureus, Klebsiella spp.
- Post-tuberculosis bronchiectasis remains significant in endemic areas
- Advanced bronchiectasis has also been reported after SARS-CoV-2 pneumonia
4. Immune / Inflammatory Disorders
- Rheumatoid arthritis, SLE, inflammatory bowel disease
- Post-transplant: chronic rejection after lung transplant; chronic graft-versus-host disease after HSCT
5. Allergic Bronchopulmonary Aspergillosis (ABPA)
- Occurs in patients with asthma or cystic fibrosis
- Hyperimmune Th2 response to Aspergillus fumigatus
- High serum IgE, serum anti-Aspergillus antibodies, intense eosinophilic airway inflammation, mucus plugs → bronchiectasis
6. Idiopathic
- Up to 50% of cases - dysfunctional host immunity to infectious agents → chronic inflammation
Pathogenesis
Two intertwined, mutually reinforcing processes:
Obstruction ↔ Chronic Infection (vicious cycle)
Obstruction → impaired secretion clearance → infection
↑ ↓
peribronchial fibrosis ←— inflammatory damage to bronchial wall
↑ ↓
traction dilation ←——— accumulating exudate distends airways
- Either process can be the initiator
- Obstruction (e.g., foreign body) → impaired drainage → bacterial superinfection → inflammatory wall destruction → irreversible dilation
- Infection (e.g., necrotizing pneumonia) → poor secretion clearance → obstruction → peribronchial fibrosis and traction → full-blown bronchiectasis
- In cystic fibrosis: CFTR defect → thick viscous secretions → obstructed mucociliary clearance → chronic bacterial infections → bronchial wall destruction + smaller bronchioles obliterated by fibrosis (bronchiolitis obliterans)
- In primary ciliary dyskinesia: dynein mutations → ciliary dysfunction → same sequence of obstruction and recurrent infection
Morphology
Gross
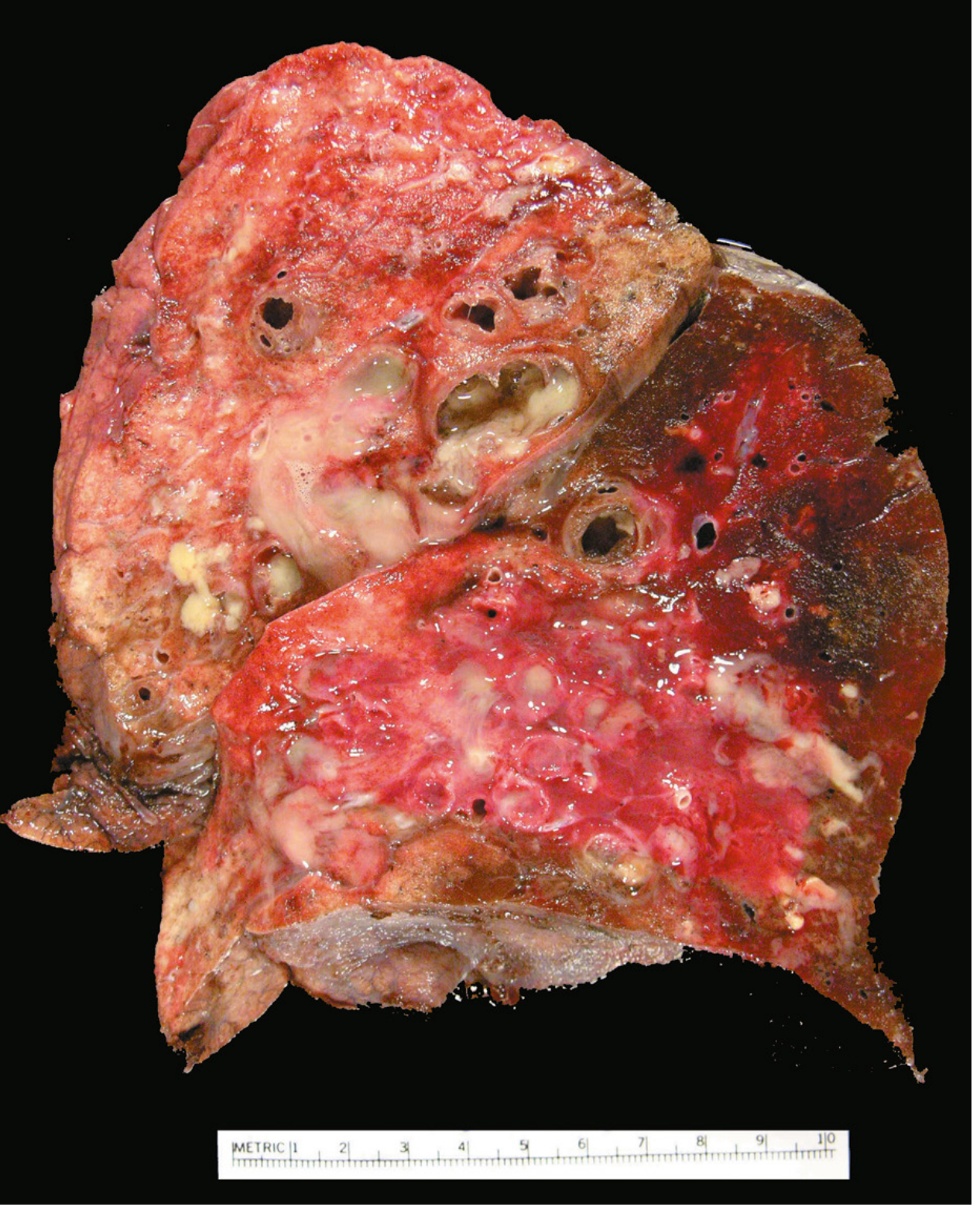
Fig. 15.12 from Robbins & Cotran: Bronchiectasis in a cystic fibrosis patient. Cut surface shows grossly dilated bronchi packed with mucopurulent secretions extending near the pleural surface.
- Lower lobes bilaterally - particularly vertical (gravity-dependent) air passages
- Airways dilated up to 4 times normal size
- Dilated bronchi visible almost to pleural surface (normally bronchioles cannot be seen beyond 2-3 cm from pleura)
- Cut surface: dilated bronchi appear cystic, filled with mucopurulent secretions
- Localized when due to tumor or foreign body
Microscopic
| Stage | Findings |
|---|---|
| Active/severe | Intense acute and chronic inflammatory exudate in bronchial/bronchiolar walls; desquamation of epithelium; extensive ulceration |
| Chronic | Squamous metaplasia of remaining epithelium (further impairs mucociliary clearance); fibrosis of bronchial and bronchiolar walls; peribronchiolar fibrosis |
| Severe/necrotic | Destruction of bronchial/bronchiolar walls → abscess cavities |
| Healing | Epithelial regeneration may occur, but dilation and scarring usually persist permanently |
Microbiology of Sputum Cultures
| Organism | Frequency |
|---|---|
| Haemophilus influenzae | ~50% |
| Pseudomonas aeruginosa | 12-30% |
| Staphylococci, streptococci, pneumococci | Common |
| Enteric organisms, anaerobes | Particularly in severe/chronic cases |
| Nontuberculous mycobacteria | Significant minority |
| Aspergillus fumigatus | In ABPA - hyphae visible on special stains within dilated bronchi |
The bronchiectatic microenvironment (inflamed, mucoid, sometimes anaerobic) supports colonization by relatively few but persistent microbial species.
Clinical Features
- Severe persistent cough with expectoration of mucopurulent, foul-smelling sputum (sometimes blood-stained)
- Hemoptysis - may occasionally be massive
- Dyspnea, orthopnea, rhinosinusitis
- Paroxysms of cough are worst in the morning on rising (change in position causes pooled pus/secretions to drain into bronchi)
- Symptoms are often episodic, precipitated by upper respiratory infections or new pathogens
Complications
| Complication | Notes |
|---|---|
| Obstructive respiratory insufficiency | Hypoxemia, hypercapnia, cyanosis |
| Pulmonary hypertension → cor pulmonale | From chronic hypoxemia |
| Brain abscess | Hematogenous spread from infected lung |
| Amyloidosis | From chronic infection/inflammation |
With modern antibiotics and physical therapy (postural drainage), life expectancy has almost doubled, and severe complications (cor pulmonale, brain abscess, amyloidosis) occur far less frequently than in the past.
Key Concept Summary
| Feature | Detail |
|---|---|
| Definition | Permanent bronchial dilation from destruction of smooth muscle and elastic tissue |
| Always secondary to | Obstruction and/or chronic infection |
| Core mechanism | Obstruction ↔ infection vicious cycle |
| Distribution | Lower lobes bilaterally; extends to pleural surface |
| Hallmark symptom | Copious foul-smelling purulent sputum; worse in morning |
| Key associations | CF, primary ciliary dyskinesia, Kartagener syndrome, ABPA, immunodeficiency, post-TB |
| Key organisms | H. influenzae, P. aeruginosa |
| Microscopic hallmark | Transmural inflammation, epithelial desquamation/ulceration, squamous metaplasia, fibrosis |
Sources: Robbins & Cotran Pathologic Basis of Disease, pp. 642-643 | Robbins & Kumar Basic Pathology, pp. 455-456
Small cell carcinoma of lung
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Your credits are exhausted. You can purchase a top-up or subscribe to a plan to continue using Orris. If you're already subscribed, your daily credits will reset tomorrow.
Make a study guide on lung cancers
Your credits are exhausted. You can purchase a top-up or subscribe to a plan to continue using Orris. If you're already subscribed, your daily credits will reset tomorrow.
This is a shared conversation. Sign in to Orris to start your own chat.