34 year female tabassum gori Kco post vsd closure at age of 14 yrs (2005) + severe pah Kco hypothyroidism since 9 yrs on rx (tab thyronorm 75mcg po 1-0-0 Patient is admitted with complaint of fever with chills and rigors with bl lowerlimb swelling and pedal edema on and off Patient refered for polycythemia and need of phlebotomy 14/6/22 Hb -18,rbc-7.13 M 26/7/22 Hb 20.6 06/02/23-cbc -19.5 /7.6/242 hct 57.2 01/09/24 cbc- 23.8/6.9/194 11/09/24 Cbc-18.8/6.5. Inr -2.98 04-04-26 Cbc -21.2/5.06/183 02-05-26 Cbc- 21.1/5.02/165. Hct-66.5 Rbc-7.49 Pt/Inr/Aptt -47.7/3.77/46.6 Ps- NORMOCYTIC NORMOCHROMIC RBCS WITH ANISOPOIKILOCYTOSIS ,MACROCYTES,FEW OVALOCYTES RDW-21.6 Tsh-2.971 Patient is in regular follow up In cardiology with spo2-70% on ra so now should we do phlebotomy for this patient
phlebotomy Eisenmenger syndrome polycythemia
phlebotomy indications Eisenmenger syndrome polycythemia secondary cyanotic congenital heart disease guidelines 2024
https://www.ahajournals.org/doi/10.1161/CIR.0000000000001402
Clinical Assessment: Phlebotomy for Tabassum Gori
Patient Summary
| Parameter | Detail |
|---|---|
| Age/Sex | 34F |
| Cardiac history | VSD closure 2005 (age 14) → Severe PAH (likely Eisenmenger physiology/post-op PAH) |
| Current SpO₂ | 70% on room air |
| Latest CBC (02/05/26) | Hb 21.1, RBC 7.49, HCT 66.5, Plt 165, RDW 21.6 |
| Coagulation | PT 47.7 / INR 3.77 / APTT 46.6 |
| PS | Normocytic normochromic + anisopoikilocytosis, macrocytes, ovalocytes |
| TSH | 2.971 (euthyroid on Thyronorm 75mcg) |
Should You Do Phlebotomy?
2025 ACC/AHA Guideline Position on Phlebotomy in Cyanotic ACHD
"Phlebotomy may be considered in patients with refractory symptoms of hyperviscosity syndrome after adequate hydration with oral or IV fluid and adequate correction of iron deficiency."
"Another indication for phlebotomy may be if the patient's hematocrit level is >65% [preoperatively for autologous blood transfusion]."
"Prophylactic phlebotomy should be avoided — it exacerbates iron deficiency and increases stroke risk. It should NOT be used as primary treatment in patients with hyperviscosity symptoms or in asymptomatic patients with elevated hematocrit, as the effect on hematocrit is transient."*
Applying to This Patient
✅ Criteria Met for Phlebotomy
- HCT 66.5% — crosses the >65% threshold
- Hb 21.1 g/dL — severely elevated
- Symptomatic: Fever, chills, rigors, bilateral lower limb edema — active clinical deterioration
- SpO₂ 70% on RA — severe chronic hypoxemia driving erythrocytosis
⚠️ Critical Concerns Before/During Phlebotomy
1. COAGULOPATHY — Most Urgent Red Flag
- INR 3.77 (current) — severely elevated
- PT 47.7s, APTT 46.6s — both prolonged
- This is characteristic of the intrinsic hemostatic defect in Eisenmenger syndrome: thrombocytopenia tendency, dysfunctional platelets, and coagulation factor depletion
- Phlebotomy in this coagulopathic state carries significant bleeding risk
- Action: Investigate the cause of INR elevation — is she on anticoagulants? Warfarin? If so, what's the indication? Hepatic dysfunction from chronic right heart failure? Correct before phlebotomy if possible, or minimize volume removed
2. IRON DEFICIENCY — Must Rule Out First
- RDW 21.6 (markedly elevated) — strongly suggests mixed picture: secondary erythrocytosis + iron deficiency
- Peripheral smear shows macrocytes + ovalocytes — macrocytes could indicate B12/folate deficiency or dyserythropoiesis; ovalocytes can be seen in iron deficiency
- Paradox: HCT appears high (66.5%) but RDW elevation + morphological changes suggest relative iron deficiency
- Iron-deficient erythrocytes are less deformable, worsening hyperviscosity even at a "normal" HCT
- Iron deficiency in Eisenmenger is associated with worse outcomes
- Action: Check serum iron, ferritin, transferrin saturation BEFORE phlebotomy. If iron-deficient, repleting iron (carefully, with monitoring) may actually reduce HCT and symptoms more safely than phlebotomy
3. HYDRATION STATUS
- Rule out dehydration as a cause of falsely elevated HCT
- Fever + rigors suggest possible infection/sepsis → dehydration likely
- Action: Adequate oral/IV hydration first, recheck HCT after rehydration
4. ACTIVE INFECTION
- Current admission: fever, chills, rigors → sepsis workup is mandatory
- Source of infection? Blood cultures, urine culture, echo to rule out infective endocarditis (high risk in this patient with structural heart disease)
- Do not perform elective phlebotomy during active sepsis — stabilize infection first
5. THROMBOCYTOPENIA TREND
- Platelets declining: 242 (2023) → 194 (2024) → 165 (2026)
- Below 200 and trending down — platelet function is also abnormal in Eisenmenger
- Combined with INR 3.77 → bleeding risk from phlebotomy is substantial
Protocol if Phlebotomy is Decided
- Volume: Remove 250–500 mL (not more) — minimum that achieves symptomatic relief
- Simultaneous volume replacement: Isotonic saline 250–500 mL (use 5% dextrose only if in heart failure to avoid fluid overload)
- Target HCT: Reduce to just below 65%, not normal range — do NOT aggressively drop HCT as this will worsen oxygen delivery
- Do NOT repeat routinely — each phlebotomy worsens iron deficiency and increases stroke risk
- After phlebotomy, check iron stores and replace iron if deficient
Overall Recommendation
| Step | Action |
|---|---|
| 1. Now | Sepsis workup (blood cultures × 2, echo, urine C/S), IV fluids for hydration |
| 2. Urgent labs | Serum iron, ferritin, TIBC/transferrin saturation; LFTs (for cause of raised INR); B12/folate |
| 3. Coagulopathy | Clarify INR 3.77 — is she on warfarin? Check if liver failure contributing. Address before phlebotomy |
| 4. If iron-deficient | Iron repletion (oral initially, IV if severe), monitor HCT response — may avoid phlebotomy |
| 5. Phlebotomy | Perform only when: (a) infection controlled, (b) coagulopathy addressed/understood, (c) iron-replete or clinically urgent with HCT >65% AND symptomatic hyperviscosity, with simultaneous 250–500 mL isotonic replacement |
| 6. Avoid | Routine/prophylactic phlebotomy, aggressive HCT reduction, phlebotomy without volume replacement |
A Note on SPO₂ 70%
- Optimizing PAH therapy (PDE5 inhibitors, endothelin receptor antagonists, prostanoids)
- Identifying if there is a residual/recurrent shunt
- Ensuring iron replete state
Change your management as pt inr is 24.7/1.84
phlebotomy safety INR 1.84 coagulopathy Eisenmenger polycythemia venesection bleeding risk
Revised Management with Corrected Coagulation: PT 24.7s / INR 1.84
What INR 1.84 Means in This Context
| Parameter | Value | Interpretation |
|---|---|---|
| PT | 24.7 sec | Mildly prolonged (normal ~11–13.5 sec) |
| INR | 1.84 | Mildly elevated — subtherapeutic anticoagulation range |
| APTT | 46.6 sec | Mildly prolonged (normal ~25–35 sec) |
- Reduced clotting factor synthesis (chronic liver congestion from right heart failure)
- Qualitative platelet dysfunction
- Reduced von Willebrand factor high-molecular-weight multimers
- Paradoxically, also at risk for both thrombosis AND bleeding
Revised Management Plan
Phlebotomy Decision: PROCEED — with conditions
- ✅ HCT 66.5% → above the >65% threshold
- ✅ Hb 21.1 g/dL → severely elevated
- ✅ Active symptoms (bilateral LL edema, fever, clinical deterioration)
- ✅ INR 1.84 — inherent Eisenmenger coagulopathy, not a contraindication
- ✅ SpO₂ 70% — erythrocytosis is compensatory but HCT >65% is now also a thrombohemorrhagic risk
Stepwise Protocol
Step 1 — Before Phlebotomy (Do These First)
| Priority | Action | Reason |
|---|---|---|
| Urgent | IV fluids (isotonic saline 500 mL) | Rule out dehydration as cause of elevated HCT; also pre-load before phlebotomy |
| Urgent | Blood cultures × 2, sepsis workup | Active fever/chills/rigors — rule out infective endocarditis (high risk) |
| Urgent | Echo (TTE/TEE) | Rule out IE vegetations, assess PAH severity, RV function |
| Same day | Serum iron, ferritin, TIBC | RDW 21.6 strongly suggests co-existing iron deficiency — critical to establish |
| Same day | LFTs + albumin | Mild PT prolongation — check hepatic synthetic function (congested liver in PAH) |
| Same day | B12/folate | Macrocytes on PS — rule out deficiency contributing to high Hb/RDW |
| Same day | Recheck HCT after hydration | If HCT drops to <65% after fluids, phlebotomy may be deferred |
Step 2 — Phlebotomy Technique
- Volume: Remove 250–500 mL (single unit) — start with 250 mL given mild coagulopathy
- Simultaneous replacement: Isotonic saline 250–500 mL IV, run concurrently (not after)
- If in heart failure: Use 5% dextrose instead of saline to avoid volume overload
- Target: Reduce HCT to just below 65% — do NOT target normal HCT; over-reduction worsens oxygen delivery at SpO₂ 70%
- Setting: Monitored bed, SpO₂ and BP monitoring throughout
- Do NOT repeat prophylactically — only repeat if symptoms of hyperviscosity recur AND iron replete AND HCT again >65%
Step 3 — Iron Deficiency Management (Mandatory)
- RDW 21.6 + macrocytes + ovalocytes on smear → this patient almost certainly has coexisting iron deficiency masked by high absolute Hb
- Iron deficiency in Eisenmenger = independent predictor of adverse outcomes
- Repeated phlebotomy without iron repletion → worsens deformability of RBCs → paradoxically increases hyperviscosity symptoms despite lower HCT
- If ferritin is low/transferrin saturation <20%: oral iron (ferrous sulfate 200mg BD) — titrate carefully; monitor for rapid HCT rise
- If severe deficiency: IV iron (Ferric carboxymaltose) with cardiology supervision
Step 4 — Address the Active Presentation
| Issue | Management |
|---|---|
| Fever/chills/rigors | Blood cultures; empiric antibiotics pending cultures if sepsis criteria met |
| Bilateral LL swelling/pedal edema | Likely right heart failure from PAH — optimize diuresis (furosemide), avoid aggressive diuresis (worsens dehydration/HCT) |
| INR 1.84 | Expected in Eisenmenger; do NOT anticoagulate — 2025 ACC/AHA: "Adults with Eisenmenger syndrome should not be routinely prescribed oral anticoagulation given the high bleeding risk and lack of long-term survival benefit" |
Step 5 — Long-Term PAH Optimization
- Current PAH medications? If not on targeted therapy → start (PDE5i: sildenafil/tadalafil; ERA: bosentan/macitentan)
- 6-minute walk test + NT-proBNP + functional class assessment for risk stratification
- Heart-lung transplant evaluation if refractory
Summary: What Changed with INR 1.84
| Before (INR 3.77 assumed) | After (INR 1.84 confirmed) |
|---|---|
| Coagulopathy was a major contraindication | INR 1.84 = expected intrinsic defect, NOT a contraindication |
| Phlebotomy deferred until INR corrected | Phlebotomy can proceed after hydration + sepsis stabilization |
| Possible need for FFP before venesection | FFP not required |
| Bleeding risk high | Bleeding risk moderate (inherent in Eisenmenger, manageable with proper technique) |
Indication of phlebotomy in secondary polycythemia
indications phlebotomy secondary polycythemia erythrocytosis guidelines 2024 2025
Indications for Phlebotomy in Secondary Polycythemia
The Fundamental Distinction
| Type | Erythrocytosis | Role of High RBC Mass | Phlebotomy? |
|---|---|---|---|
| Physiologically INAPPROPRIATE | EPO-secreting tumor, post-renal transplant, testosterone therapy, Cushing's | Not needed for O₂ delivery | Yes — aggressive, target HCT <45% |
| Physiologically APPROPRIATE | Cyanotic CHD, COPD, high altitude, sleep apnea, chronic hypoxia | Life-sustaining compensatory mechanism | Restricted — only specific indications |
A. Physiologically INAPPROPRIATE Secondary Polycythemia
Indications — Liberal
- Symptomatic hyperviscosity — headache, dizziness, blurred vision, tinnitus, plethora, fatigue
- HCT persistently >45% — target HCT 42–46% with regular phlebotomy
- Prior to elective surgery — mandatory phlebotomy to reduce thrombotic/bleeding perioperative risk
- Thrombotic risk reduction — these patients have increased risk of CVA, MI, DVT, PE
- Temporary relief while definitive treatment of underlying cause is arranged (e.g. tumor resection)
B. Physiologically APPROPRIATE Secondary Polycythemia
Indications — Restrictive (all 3 ideally present)
1. Symptomatic Hyperviscosity Syndrome
- Visual disturbances, blurred vision
- Severe headache
- Dizziness, vertigo, tinnitus
- Paresthesia, digital numbness
- Profound fatigue, reduced concentration
- ⚠️ Always rule out dehydration first — dehydration falsely elevates HCT and mimics hyperviscosity symptoms
2. HCT Threshold
- HCT >65% — at this level, compensatory benefit plateaus and hyperviscosity overrides O₂ delivery benefit
- Below 65%: viscosity is still within tolerable range; O₂ delivery benefit outweighs risk of phlebotomy
3. Iron-Replete State
- Must confirm iron sufficiency before proceeding
- Iron deficiency → rigid, non-deformable RBCs → worsened viscosity at same or even lower HCT
- Phlebotomy in iron-deficient patient = worsening of the very problem you're trying to treat
- Check ferritin, serum iron, TIBC before each session
Additional Specific Indications (Any Type of Secondary Polycythemia)
| Indication | Detail |
|---|---|
| Pre-operative phlebotomy | HCT >65% before non-cardiac elective surgery; reduce to just below 65% with isovolumetric replacement |
| Acute hyperviscosity crisis | Sudden neurological deterioration, visual loss, acute stroke-like symptoms with HCT >65% |
| Autologous blood banking | Pre-op collection for anticipated surgical blood loss |
Absolute Contraindications / When NOT to Phlebotomize
| Contraindication | Reason |
|---|---|
| Asymptomatic patient with high HCT (physiologically appropriate) | Removes the compensatory mechanism; worsens tissue hypoxia |
| Iron-deficient state (not corrected) | Makes hyperviscosity worse post-phlebotomy |
| Active dehydration | HCT falsely elevated; rehydration alone will correct |
| Severe thrombocytopenia (<50,000) without correction | Bleeding risk |
| Routine/prophylactic phlebotomy | No survival benefit; causes iron deficiency and increases stroke risk (2025 ACC/AHA: Class III — No Benefit) |
| COPD with SpO₂ <88% on room air, not otherwise meeting criteria | Polycythemia is compensatory; reducing it worsens exercise tolerance |
Phlebotomy Technique When Indicated
| Parameter | Recommendation |
|---|---|
| Volume per session | 250–500 mL (minimum that achieves symptomatic relief) |
| Volume replacement | Equal volume isotonic saline simultaneously (5% dextrose if heart failure) |
| Target HCT | Just below 65% (appropriate erythrocytosis) / <45% (inappropriate erythrocytosis) |
| Frequency | Only as needed for symptoms; not scheduled/routine |
| Monitoring | SpO₂, BP, HR throughout |
| Post-phlebotomy | Check iron stores; replete if deficient |
Summary Table
| Cause | Phlebotomy Indication | Target HCT |
|---|---|---|
| EPO-secreting tumor | Symptomatic OR HCT >45% | <45% |
| Post-renal transplant | Symptomatic OR HCT >52% | <45% |
| Testosterone-induced | HCT >54% (AUA guideline) | <54% |
| Cyanotic CHD / Eisenmenger | HCT >65% + symptomatic hyperviscosity + iron replete | Just below 65% |
| COPD/sleep apnea | Rarely; only if HCT >65% + refractory hyperviscosity symptoms | Just below 65% |
| High altitude | Treat cause (descent/O₂); phlebotomy rarely needed | Just below 65% |
Thrombophillia
Thrombophilia
Definition
Classification
I. Inherited (Primary) Thrombophilias
A. Loss-of-Function Mutations (Anticoagulant Deficiencies)
| Defect | Mechanism | Prevalence in General Population | VTE Risk (Relative) |
|---|---|---|---|
| Antithrombin III deficiency | ↓ inhibition of thrombin, Xa, IXa | 1 in 2,000–5,000 | 5–50× ↑ |
| Protein C deficiency | ↓ inactivation of FVa and FVIIIa | 1 in 200–500 | 3–15× ↑ |
| Protein S deficiency | ↓ cofactor for Protein C | 1 in 200–500 | 5–10× ↑ |
Protein S is the cofactor for Protein C. Both are vitamin K–dependent. Important clinical consequence: warfarin-induced skin necrosis — warfarin drops Protein C and S faster than procoagulant factors (II, VII, IX, X), creating a transient prothrombotic window → always bridge with heparin 2–3 days before warfarin loading in these patients. (Quick Compendium of Clinical Pathology, 5th Ed.)
B. Gain-of-Function Mutations (Procoagulant Excess)
1. Factor V Leiden (FVL) — Most Common Inherited Thrombophilia
- Mutation: c.1691G>A in FV gene → Arg506Gln substitution
- Mechanism: APC (activated protein C) normally cleaves FVa at Arg506 to inactivate it. The Gln substitution resists APC cleavage → FVa persists → 10-fold prolongation of APC inactivation → excess thrombin generation
- Prevalence: 3–5% in White/Northern European populations; nearly absent in African and Asian populations
- VTE risk:
- Heterozygous: 5–10× increased relative risk; mean onset age 44 years
- Homozygous: 10–50× increased; mean onset age 31 years
- Represents: ~25% of idiopathic VTE, 30–50% of recurrent VTE
- Note: Does NOT increase arterial thrombosis risk
2. Prothrombin G20210A Mutation
- Mutation: G→A substitution at nucleotide 20210 of prothrombin gene
- Mechanism: Gain-of-function → elevated plasma prothrombin levels → excess thrombin
- Prevalence: 1–6% in White populations; rare in other groups
- Found in: 3–8% of all VTE patients; 2–3× increased VTE risk
C. Other Inherited Procoagulant States
- Elevated Factor VIII — significant independent VTE risk factor; often genetically determined
- Factor IX Padua — X-linked thrombophilia (gain-of-function FIX mutation)
- Prothrombin Yukuhashi — rare missense mutation; impaired antithrombin inhibition of thrombin
- Elevated Factors VII, IX, XI, fibrinogen, vWF, thrombin-activatable fibrinolysis inhibitor (TAFI)
- ↓ Tissue Factor Pathway Inhibitor (TFPI)
- Rare: Heparin cofactor II deficiency, dysfunctional thrombomodulin, fibrinolytic disorders (hypoplasminogenemia, PAI-1 excess, dysplasminogenemia)
II. Acquired (Secondary) Thrombophilias
| Disorder | Notes |
|---|---|
| Antiphospholipid Syndrome (APS) | Most important acquired thrombophilia; causes both arterial AND venous thrombosis |
| Malignancy | Trousseau syndrome; tissue factor overexpression |
| Oral contraceptives / HRT / Pregnancy | Estrogen → ↑ procoagulant factors, ↓ Protein S |
| Myeloproliferative neoplasms (PV, ET) | Platelet activation + JAK2 mutation → splanchnic, cerebral vein thrombosis |
| Nephrotic syndrome | Urinary loss of antithrombin III |
| Hyperhomocysteinemia | Endothelial injury → arterial + venous thrombosis |
| Immobility, surgery, trauma | Virchow's triad — stasis predominant |
| Obesity, IBD, autoimmune disorders | Chronic inflammation → hypercoagulable state |
| Heparin-induced thrombocytopenia (HIT) | Paradoxical thrombosis despite anticoagulation |
Antiphospholipid Syndrome (APS) — Key Acquired Thrombophilia
- Antibodies: Lupus anticoagulant (LA), anti-cardiolipin IgG/IgM, anti-β2 glycoprotein I IgG/IgM
- "Triple positive" (LA + 2 positive IgG antiphospholipid antibodies) = highest thrombotic risk
- Unique feature: Causes BOTH arterial AND venous thrombosis (unlike primary thrombophilias)
- Treatment: Warfarin preferred (not DOACs) for secondary prevention in APS, especially triple-positive; target INR 2.0–3.0
- Obstetric APS: Recurrent pregnancy loss, fetal loss, pre-eclampsia
Clinical Manifestations
| Feature | Details |
|---|---|
| Typical site | DVT of lower extremities, PE — most common |
| Unusual sites | Splanchnic vein thrombosis (mesenteric, portal, hepatic/Budd-Chiari), cerebral venous sinus thrombosis, retinal vein |
| Arterial thrombosis | NOT characteristic of inherited thrombophilias; suggests APS, hyperhomocysteinemia, myeloproliferative disorder, or paradoxical embolism via PFO |
| Age of onset | Typically early adulthood; family history often positive |
| Recurrence | High — unprovoked VTE is a chronic disease without prophylaxis |
| Pregnancy complications | Recurrent pregnancy loss (especially APS); peripartum DVT/PE |
Diagnosis — Who and When to Test
Who to Test
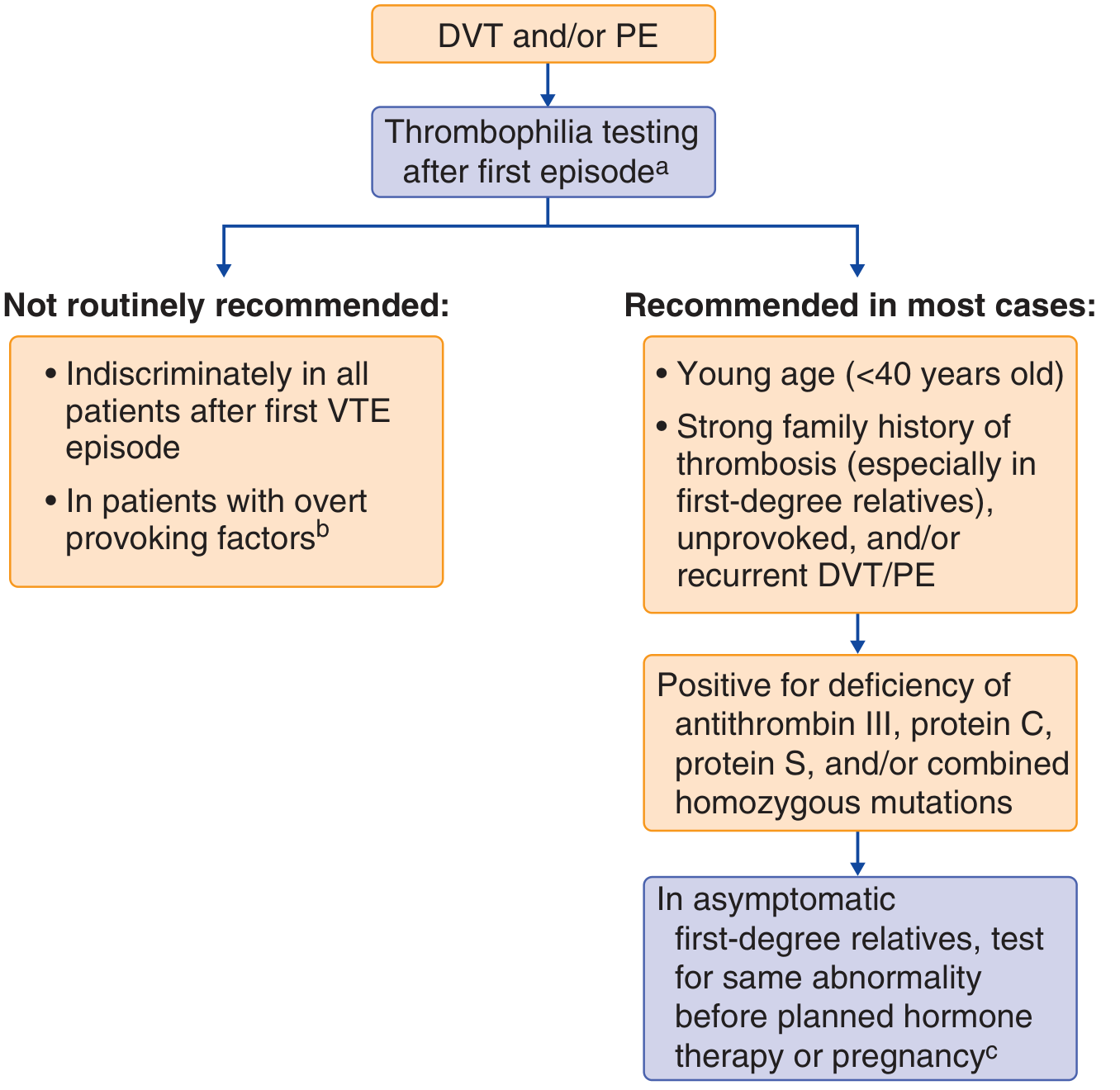
- Age <40 at first VTE
- Unprovoked VTE (no clear precipitant)
- Recurrent VTE
- Strong family history of thrombosis (first-degree relatives)
- Unusual site of thrombosis (cerebral, splanchnic)
- Pregnancy-associated thrombosis
- All patients after first VTE indiscriminately
- VTE with overt provoking factors (major surgery, trauma, prolonged immobility)
- Arterial thrombosis alone (primary thrombophilias are not the cause)
When to Test — Timing Is Critical
| Condition | Effect on Tests |
|---|---|
| During acute thrombosis | Consumes antithrombin, Protein C, Protein S → false deficiency |
| On heparin | Lowers functional antithrombin activity |
| On warfarin | Lowers Protein C and Protein S levels → false deficiency |
| On DOACs | Can falsely elevate APC resistance ratio; interfere with lupus anticoagulant testing |
| During pregnancy/estrogen use | Affects Protein S levels |
Laboratory Tests
| Test | Detects |
|---|---|
| Antithrombin III activity (functional) | AT III deficiency |
| Protein C activity (functional) + antigen | Protein C deficiency (Type I vs II) |
| Protein S free antigen + total antigen | Protein S deficiency |
| APC Resistance assay (APCR ratio) | Phenotypic screen for FV Leiden |
| Factor V Leiden PCR (c.1691G>A) | Genotypic confirmation; unaffected by anticoagulants |
| Prothrombin G20210A PCR | Prothrombin mutation |
| Lupus anticoagulant panel (dRVVT, SCT) | APS |
| Anticardiolipin IgG/IgM | APS |
| Anti-β2 glycoprotein I IgG/IgM | APS |
| Homocysteine (fasting) | Hyperhomocysteinemia |
| Factor VIII activity | Elevated FVIII as risk factor |
| JAK2 V617F | Myeloproliferative neoplasm |
Treatment
Acute VTE
- Initial management is the same regardless of thrombophilia genotype
- DOACs (rivaroxaban, apixaban, edoxaban, dabigatran) — first-line; at least as effective as warfarin with fewer bleeds
- Exceptions where warfarin is preferred:
- Antiphospholipid syndrome (especially triple-positive)
- Prosthetic heart valves
Duration of Anticoagulation
| Scenario | Duration |
|---|---|
| Provoked VTE (transient risk factor) | 3–6 months |
| Unprovoked first VTE | Minimum 6–12 months; consider indefinite |
| Recurrent VTE (≥2 events) | Indefinite anticoagulation regardless of thrombophilia status |
| High-risk thrombophilia (AT III deficiency, Protein C/S deficiency, homozygous FVL, APS) | Consider indefinite after first unprovoked VTE |
Prophylaxis in High-Risk Situations
- Peripartum period
- Major surgery / prolonged immobilization
- Long-haul travel
- Hospitalization
Summary: Risk Comparison
| Thrombophilia | Relative Risk of VTE | Prevalence |
|---|---|---|
| Antithrombin III deficiency | 5–50× | Very rare |
| Protein C deficiency | 3–15× | 1 in 200–500 |
| Protein S deficiency | 5–10× | 1 in 200–500 |
| FV Leiden (heterozygous) | 5–10× | 3–5% (White) |
| FV Leiden (homozygous) | 50–100× | Rare |
| Prothrombin G20210A | 2–3× | 1–6% (White) |
| Antiphospholipid syndrome | 5–10× (venous + arterial) | 1–5% |
More detailed also explain about factor 2 deficiency
Thrombophilia — Comprehensive Detail
Definition & Core Concept
"Similar to seizures, patients have a varying threshold for clotting." (Tintinalli's Emergency Medicine)
Virchow's Triad — Pathophysiological Basis
- Stasis — immobility, CHF, post-surgical, atrial fibrillation
- Endothelial injury — atherosclerosis, trauma, surgery, hyperhomocysteinemia
- Hypercoagulability — inherited or acquired thrombophilia
PART I — INHERITED (PRIMARY) THROMBOPHILIAS
Physiologic Anticoagulant Systems (What Goes Wrong)
- ~2,000–4,000× for thrombin
- ~500–1,000× for Factor Xa
- ~1,000,000× for Factor IXa
A. ANTITHROMBIN III DEFICIENCY
| Feature | Detail |
|---|---|
| Prevalence | 1 in 10,000 general population; ~1% of familial VTE |
| Inheritance | Autosomal dominant, high penetrance |
| Heterozygote AT levels | 40–60% of normal → full clinical manifestations |
| VTE risk | 5–50× increased — highest risk of all inherited thrombophilias |
Types
| Type | AT Activity | AT Antigen | Notes |
|---|---|---|---|
| Type I | ↓ | ↓ (proportionate) | Quantitative deficiency; frameshift/nonsense mutations |
| Type II RS | ↓ | Normal | Reactive site defect — mutations near thrombin binding site |
| Type II HBS | ↓ | Normal | Heparin binding site defect → heparin resistance; low thrombotic risk as heterozygote |
| Type II PE | ↓ | Normal | Pleiotropic effect — affects both sites |
Key Clinical Points
- Homozygous AT III deficiency is incompatible with life (intrauterine thrombosis) — except Type II HBS heterozygotes who survive
- Most common presentation: DVT lower extremity + PE; also retinal, mesenteric, splenic vein thrombosis
- Heparin resistance — heparin works through AT III; if AT III is absent/low, heparin cannot exert full anticoagulant effect → escalating heparin doses required → use AT III concentrate (recombinant or plasma-derived) as adjunct
- Acquired AT III deficiency: DIC, liver disease, nephrotic syndrome (urinary loss), post-thrombosis, asparaginase chemotherapy, heparin therapy (↓ up to 20%)
- Newborns have physiologically low AT III levels — reaches adult levels by 6 months
Diagnosis
- Functional (chromogenic) assay — preferred; detects all types including Type II
- Antigen assay (ELISA/nephelometry) — to distinguish Type I from Type II
- Never rely on antigen alone — Type II has normal antigen but low function
Treatment
- Acute VTE: standard anticoagulation; if heparin-resistant → AT III concentrate IV
- Long-term: DOACs or warfarin; indefinite anticoagulation after first unprovoked VTE recommended given very high recurrence risk
B. PROTEIN C DEFICIENCY
| Feature | Detail |
|---|---|
| Prevalence | 1 in 200–500; found in 2–5% of VTE patients |
| Inheritance | Autosomal dominant |
| Function | Inactivates FVa and FVIIIa after activation by thrombin on endothelium |
| VTE risk | 3–15× increased |
Types
| Type | PC Activity | PC Antigen |
|---|---|---|
| Type I | ↓ | ↓ (proportionate) — quantitative deficiency |
| Type II | ↓ | Normal — qualitative/functional defect |
Key Clinical Points
- Neonatal purpura fulminans — homozygous or doubly heterozygous Protein C deficiency → widespread, potentially fatal thrombosis in newborns; a hematological emergency requiring Protein C concentrate
- Warfarin-induced skin necrosis (WISN) — the highest-risk thrombophilia for this complication:
- Warfarin inhibits vitamin K–dependent proteins. Protein C has shortest half-life (~6–8 hours) → drops first, before procoagulant factors (II, VII, IX, X)
- Creates a transient prothrombotic window of ~3–5 days after starting warfarin
- Dermal venous thrombosis → painful skin necrosis, typically over fat-rich areas (breast, buttocks, thighs, abdomen)
- Prevention: Always overlap with heparin (UFH or LMWH) for ≥5 days when initiating warfarin; avoid large loading doses
- Management if occurs: Switch to heparin acutely; rivaroxaban shown safe as long-term alternative; Protein C concentrate if available
- Acquired Protein C deficiency: liver disease, DIC, vitamin K deficiency, post-op, neonatal period
Diagnosis
- Functional assay preferred (chromogenic or clot-based)
- Antigen assay to distinguish Type I from Type II
- Cannot test reliably during: acute thrombosis, warfarin therapy, vitamin K deficiency (all reduce Protein C)
C. PROTEIN S DEFICIENCY
| Feature | Detail |
|---|---|
| Prevalence | ~1 in 500; found in 1–3% of VTE patients |
| Inheritance | Autosomal dominant |
| Function | Cofactor for activated Protein C; amplifies FVa/FVIIIa inactivation ~20× |
| VTE risk | 5–10× increased (but only markedly elevated when levels are far below normal range) |
Types
| Type | Free Protein S | Total Protein S | PC Activity Supported |
|---|---|---|---|
| Type I | ↓ | ↓ | ↓ |
| Type II | Normal | Normal | ↓ (functional defect) |
| Type III | ↓ | Normal | ↓ (↓ free fraction) |
Key Clinical Points
- In plasma, ~60% of Protein S is bound to C4b-binding protein (inactive); only the free form (~40%) is functionally active as Protein C cofactor
- Protein S deficiency mimics Protein C deficiency clinically — same sites of thrombosis
- Also associated with WISN (mechanism same as Protein C deficiency)
- Neonatal purpura fulminans in homozygous deficiency
- Acquired: liver disease, pregnancy, OCP/estrogen use (↑ C4b-binding protein → ↓ free fraction), Vitamin K deficiency, DIC
Diagnosis
- Free Protein S antigen — most important; reflects functionally available fraction
- Total Protein S antigen + functional assay to classify Type I/II/III
- Cannot test during: estrogen/OCP use, pregnancy, warfarin therapy, acute thrombosis
D. FACTOR V LEIDEN (FVL) — Most Common Inherited Thrombophilia
| Feature | Detail |
|---|---|
| Mutation | c.1691G>A, exon 10 of FV gene → Arg506Gln |
| Mechanism | APC cannot cleave FVa at Arg506 → FVa persists 10× longer → excess thrombin |
| Prevalence | 3–5% White/Northern European; absent in African & Asian populations |
| Inheritance | Autosomal dominant |
| Zygosity | VTE Relative Risk | Mean onset |
|---|---|---|
| Heterozygous | 5–10× | 44 years |
| Homozygous | 50–100× | 31 years |
Combined Risk (Multiplicative Effects)
| Combination | Combined Fold Risk |
|---|---|
| FVL (het) + Protein C deficiency | 25–45× |
| FVL (het) + Protein S deficiency | 25–50× |
| FVL (het) + elevated Factor VIII | 12–20× |
| FVL (het) + OCP | 8–20× |
| FVL (het) + Pregnancy | 25–40× |
Key Points
- Does NOT increase arterial thrombosis risk
- Found in 25% of idiopathic VTE, 30–50% of recurrent VTE, 20–60% of OCP-associated VTE, 8–30% of recurrent pregnancy loss
Diagnosis
- APCR phenotypic assay: ratio = APTT with APC ÷ APTT without APC; low ratio = resistance
- Modified assay (1:5 dilution in FV-depleted plasma) is more specific for FVL
- DOACs can falsely elevate APC ratio
- Molecular (PCR) testing for c.1691G>A — definitive; unaffected by anticoagulation; required to distinguish heterozygous from homozygous; critical for genetic counseling
E. PROTHROMBIN G20210A MUTATION
| Feature | Detail |
|---|---|
| Mutation | G→A at nucleotide 20210 of F2 (prothrombin) gene — 3'-UTR |
| Mechanism | Gain-of-function → ~30% increase in circulating prothrombin → excess thrombin generation |
| Prevalence | 1–6% White/European; rare in other populations |
| Inheritance | Autosomal dominant |
| VTE risk | 2–3× increased (heterozygous); higher with homozygosity |
| Frequency in VTE patients | 3–8% of all VTE; ~10% of first-episode DVT |
Key Points
- Similar clinical phenotype to FVL — predominantly venous thrombosis
- Also associated with pregnancy complications (pre-eclampsia, fetal loss, IUGR)
- Combined FVL + prothrombin mutation = very high VTE risk
- Diagnosis: PCR for G20210A — lab tests of coagulation (PT/aPTT) may be normal; prothrombin activity/antigen elevated
F. FACTOR II (PROTHROMBIN) DEFICIENCY — Hemorrhagic Disorder, NOT Thrombophilia
| Feature | Detail |
|---|---|
| Prevalence | ~1 in 2,000,000 — rarest inherited coagulation deficiency |
| Complete deficiency | Incompatible with life (animal models confirm perinatal lethality) |
| Inheritance | Autosomal recessive |
Types
| Type | FII Activity | FII Antigen | Clinical |
|---|---|---|---|
| Type I — Hypoprothrombinemia | ↓ | ↓ (proportionate) | Mucosal bleeding, hematomas, hemarthrosis |
| Type II — Dysprothrombinemia | ↓ | Normal (dysfunctional protein) | Less predictable — may be asymptomatic or have mild bleeding only |
Clinical Features
- Bleeding symptoms: mucosal bleeding (epistaxis, gum bleeding, menorrhagia), easy bruising, hematomas, hemarthrosis (joint bleeding)
- Severity correlates with residual FII activity level
- Dysprothrombinemia (Type II) is clinically milder and unpredictable
Lab Pattern
| Test | Result |
|---|---|
| PT | Prolonged |
| aPTT | Prolonged |
| Thrombin time | Normal (fibrinogen is normal) |
| Specific FII assay | Low activity (confirms) |
- Both PT and aPTT are prolonged because FII is in the common pathway (shared by intrinsic and extrinsic pathways)
- Compare: Factor VII deficiency → prolonged PT only (extrinsic); Factor VIII/IX deficiency → prolonged aPTT only (intrinsic)
Acquired Factor II Deficiency (More Common)
- Warfarin / Vitamin K deficiency — FII is vitamin K–dependent; drops with warfarin (half-life 3 days — slowest of all vitamin K–dependent factors)
- Liver disease — reduced hepatic synthesis
- DIC — consumption
- Antiphospholipid syndrome with lupus anticoagulant (LA) — rare acquired autoantibody can target prothrombin → lupus anticoagulant-hypoprothrombinemia syndrome (LAHPS) — causes bleeding despite a lupus anticoagulant (which usually causes thrombosis)
Treatment of FII Deficiency
| Option | Notes |
|---|---|
| Prothrombin Complex Concentrate (PCC) | Treatment of choice; contains FII, VII, IX, X |
| Fresh Frozen Plasma (FFP) | Alternative when PCC unavailable; larger volumes needed |
| Recombinant FII | Not currently available |
| Vitamin K | Effective only if cause is warfarin or dietary vitamin K deficiency |
PART II — ACQUIRED (SECONDARY) THROMBOPHILIAS
Antiphospholipid Syndrome (APS) — Full Detail
| Feature | Detail |
|---|---|
| Antibodies | Lupus anticoagulant (LA), anti-cardiolipin IgG/IgM, anti-β2 glycoprotein I IgG/IgM |
| Unique feature | Only thrombophilia causing both arterial AND venous thrombosis |
| Triple positive | LA + 2 positive IgG antiphospholipid antibodies = highest risk |
| Obstetric APS | Recurrent pregnancy loss, pre-eclampsia, IUGR, placental insufficiency |
Catastrophic APS (CAPS)
- Rapid multiorgan thrombosis (<1 week), high mortality
- Treat with anticoagulation + steroids + IVIG/plasmapheresis
Lab Paradox
- Lupus anticoagulant prolongs aPTT in vitro (phospholipid-dependent test) but causes thrombosis in vivo
- Cannot test lupus anticoagulant on DOACs — falsely positive results
Treatment
- Warfarin preferred over DOACs for APS (especially triple-positive) — target INR 2.0–3.0
- DOACs have shown higher recurrence rates in APS (TRAPS trial with rivaroxaban)
Full Table of Acquired Thrombophilias (Secondary Hypercoagulable States)
| Condition | Mechanism |
|---|---|
| Surgery / trauma / immobility | Stasis + endothelial injury + coagulation activation |
| Pregnancy + puerperium | ↑ procoagulant factors, ↓ Protein S, ↓ fibrinolysis, stasis |
| OCP / HRT / tamoxifen | Estrogen → ↑ FVII, FVIII, FX, fibrinogen; ↓ Protein S, AT III |
| Malignancy (Trousseau) | Tumor-derived tissue factor, mucin activates platelets/coagulation |
| Myeloproliferative neoplasms (PV, ET) | JAK2 V617F → sticky activated platelets, especially splanchnic veins |
| Paroxysmal nocturnal hemoglobinuria (PNH) | Complement-activated platelets; especially hepatic vein thrombosis |
| Nephrotic syndrome | Urinary loss of AT III, Protein C, Protein S |
| Hyperhomocysteinemia | Endothelial injury, ↑ oxidative stress; arterial + venous thrombosis |
| Heparin-induced thrombocytopenia (HIT) | PF4-heparin antibodies → platelet activation → paradoxical thrombosis |
| Antiphospholipid syndrome | Antibody-mediated activation of endothelium and coagulation |
| HIV | Endothelial inflammation, acquired Protein S deficiency |
| DIC | Paradoxically thrombotic AND hemorrhagic |
| Non-O blood group | vWF cleared more slowly → higher vWF levels → ↑ FVIII → VTE risk |
Thrombophilia Testing — Complete Framework
When to Suspect (Features)
- Age ≤45 at first VTE
- Recurrent VTE
- Family history of thrombosis (first-degree relatives)
- Unprovoked (no clear provoking factor)
- Unusual site: hepatic, portal, mesenteric, splenic, cerebral venous sinus
- Recurrent pregnancy loss / adverse obstetric outcomes
- Neonatal purpura fulminans
- Warfarin-induced skin necrosis
Testing Algorithm
When NOT to Test
- During acute thrombosis (consumes AT III, Protein C, Protein S — false deficiency)
- On warfarin (lowers Protein C, Protein S)
- On heparin (lowers AT III activity)
- On DOACs (interferes with LA testing, APCR ratio)
- Arterial thrombosis alone (primary thrombophilias rarely causative)
Optimal Timing
Treatment — Comprehensive
Acute VTE
- DOACs first-line (rivaroxaban, apixaban, edoxaban, dabigatran) — at least as effective as warfarin, fewer bleeds
- Exceptions where warfarin preferred: APS (especially triple-positive), prosthetic heart valves, mechanical heart valves
- Bridge with UFH/LMWH for ≥5 days if starting warfarin in Protein C or S deficiency
Duration of Anticoagulation
| Scenario | Duration |
|---|---|
| Provoked VTE (major transient: surgery, trauma) | 3–6 months |
| Provoked VTE (minor transient: OCP, travel) | 3–6 months, consider longer |
| First unprovoked VTE — low-risk thrombophilia (het FVL, het PT G20210A) | 6–12 months minimum; weigh bleeding vs recurrence |
| First unprovoked VTE — high-risk thrombophilia (AT III, Protein C/S deficiency, homo FVL, APS) | Indefinite anticoagulation |
| Recurrent VTE (≥2 events) | Indefinite, regardless of thrombophilia status |
Asymptomatic Relatives
- Do NOT anticoagulate prophylactically in absence of VTE history
- Counsel about: avoiding OCP/HRT, prophylaxis before surgery/pregnancy, signs of DVT/PE
- Test before planned hormone therapy or pregnancy if family member has high-risk thrombophilia
Pregnancy in Thrombophilia
- LMWH — anticoagulant of choice throughout pregnancy
- Warfarin: crosses placenta → fetal warfarin syndrome (teratogenic in 1st trimester), fetal bleeding
- DOACs: safety in pregnancy not established — avoid
- Thromboprophylaxis throughout pregnancy + 6 weeks postpartum (highest risk period) (Goldman-Cecil Medicine)
Risk Summary — All Thrombophilias
| Disorder | Type | VTE Relative Risk | Prevalence (general pop) |
|---|---|---|---|
| Antithrombin III deficiency | Inherited | 5–50× | 1 in 10,000 |
| Protein C deficiency | Inherited | 3–15× | 1 in 200–500 |
| Protein S deficiency | Inherited | 5–10× | 1 in 500 |
| Factor V Leiden (het) | Inherited | 5–10× | 3–5% (White) |
| Factor V Leiden (hom) | Inherited | 50–100× | Rare |
| Prothrombin G20210A (het) | Inherited | 2–3× | 1–6% (White) |
| FVL + PT G20210A (combined) | Inherited | Very high | Uncommon |
| Antiphospholipid syndrome | Acquired | 5–10× (art+ven) | 1–5% |
| Factor II deficiency | Inherited | — (bleeding disorder) | 1 in 2,000,000 |
| Hyperhomocysteinemia | Acquired/genetic | 2–3× | Variable |