Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND "pathophysiology"[MeSH Terms]
Searching Images
ARDS acute respiratory distress syndrome pathophysiology mechanism diagram
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Reading File
Reading File
Reading File
I now have a comprehensive picture of the ARDS mechanism from Murray & Nadel's Textbook of Respiratory Medicine. I have enough material to construct a thorough, well-cited answer. Let me now compose the full response.
Mechanism of Acute Respiratory Distress Syndrome (ARDS)
ARDS is a syndrome of acute, noncardiogenic pulmonary edema driven by severe lung inflammation, increased alveolar-capillary permeability, and profound hypoxemia. Rather than a single pathophysiologic pathway, it represents a common clinical endpoint reached by multiple injurious processes.
1. Definition & Diagnostic Context
The Berlin definition (2012) classifies ARDS by PaO₂/FiO₂ ratio on ≥5 cmH₂O PEEP:
- Mild: PaO₂/FiO₂ 200–300
- Moderate: 100–200
- Severe: <100
Onset must occur within 7 days of a known clinical insult (e.g., sepsis, pneumonia, trauma, transfusion, aspiration, pancreatitis), with bilateral opacities not fully explained by cardiac failure or fluid overload. — Murray & Nadel's Textbook of Respiratory Medicine
2. Inciting Stimuli
ARDS arises from direct (pulmonary) or indirect (extrapulmonary) insults:
| Direct | Indirect |
|---|---|
| Pneumonia | Sepsis |
| Aspiration of gastric contents | Major trauma |
| Pulmonary contusion | Pancreatitis |
| Inhalation injury | Blood product transfusion (TRALI) |
| Drowning | Burn injury |
3. Pathologic Phases
The pathology of ARDS has three overlapping, sequential stages known as diffuse alveolar damage (DAD):
Phase 1 — Exudative (Days 1–7)
- Protein-rich, exudative fluid floods the alveolar spaces
- Hyaline membranes form — composed of cellular debris, proteins, and surfactant remnants
- Widespread epithelial disruption
- Massive neutrophil infiltration of interstitium and airspaces
- Loss of surfactant → alveolar collapse and decreased compliance
Phase 2 — Proliferative (Days 7–21)
- Hyaline membranes are reorganized
- Early fibrosis appears
- Type II pneumocyte proliferation attempts epithelial repair
- Decreased neutrophil infiltration
- Reduction in pulmonary edema
Phase 3 — Fibrotic (>2–3 weeks, subset of patients)
- Established pulmonary fibrosis
- Obliteration of pulmonary capillaries
- Deposition of interstitial and alveolar collagen
- Notably, evidence of fibroproliferation (elevated N-terminal procollagen peptide III in BAL fluid) may begin as early as 24 hours after injury onset
— Murray & Nadel's Textbook of Respiratory Medicine
4. Core Mechanism: Alveolar-Capillary Barrier Disruption
The central event is increased alveolar-capillary permeability, involving both the microvascular endothelium and the alveolar epithelium.
Endothelial Injury
- Normally, the pulmonary microvascular endothelium presents a tight barrier maintained by cell-cell junctions (adherens junctions, tight junctions)
- In ARDS, inflammatory mediators (TNF-α, IL-1β, IL-8) damage these junctions
- Destruction of endothelial cadherins (key adherens junction proteins) by neutrophil elastase leads to paracellular leak
- Result: protein-rich plasma fluid escapes into the interstitium and alveolar spaces
Epithelial Injury
- Type I pneumocytes (covering ~95% of alveolar surface) are particularly vulnerable
- Their loss impairs the normal fluid-clearing function of the epithelial barrier
- Type II pneumocytes are the primary survivors and must regenerate type I cells
- Injury to type II cells impairs surfactant production, worsening alveolar instability
5. The Neutrophil as Central Effector
Neutrophils are the primary mediators of lung injury in ARDS:
-
Sequestration: Neutrophils are retained in pulmonary capillaries and recruited into alveolar spaces following exposure to chemoattractants (IL-8/CXCL8, C5a, leukotriene B4)
-
Reactive oxygen species (ROS): Oxidative burst generates superoxide, hydrogen peroxide, and hypochlorous acid — directly damaging epithelial and endothelial cells
-
Proteolytic enzymes:
- Neutrophil elastase (NE): Degrades growth factors, cytokines, and epithelial/endothelial cadherins — predisposing to alveolar flooding
- Matrix metalloproteinases (MMPs): Destroy basement membrane components
-
Neutrophil Extracellular Traps (NETs): Web-like structures of DNA, histones, myeloperoxidase, NE, and cathepsin G — released to trap pathogens. In ARDS, NETs cause endothelial damage and promote thrombus formation. BAL fluid from ARDS patients shows NET concentrations inversely correlated with PaO₂/FiO₂
-
Platelet-neutrophil complexes: Via P-selectin/PSGL-1 interactions, platelets and neutrophils form complexes that amplify ROS and NET release, driving cell injury through necroptosis and pyroptosis
Importantly, ARDS can occur in profoundly neutropenic patients, implicating alveolar macrophages as an alternative effector — Murray & Nadel's
6. Cytokine Storm & Macrophage Activation
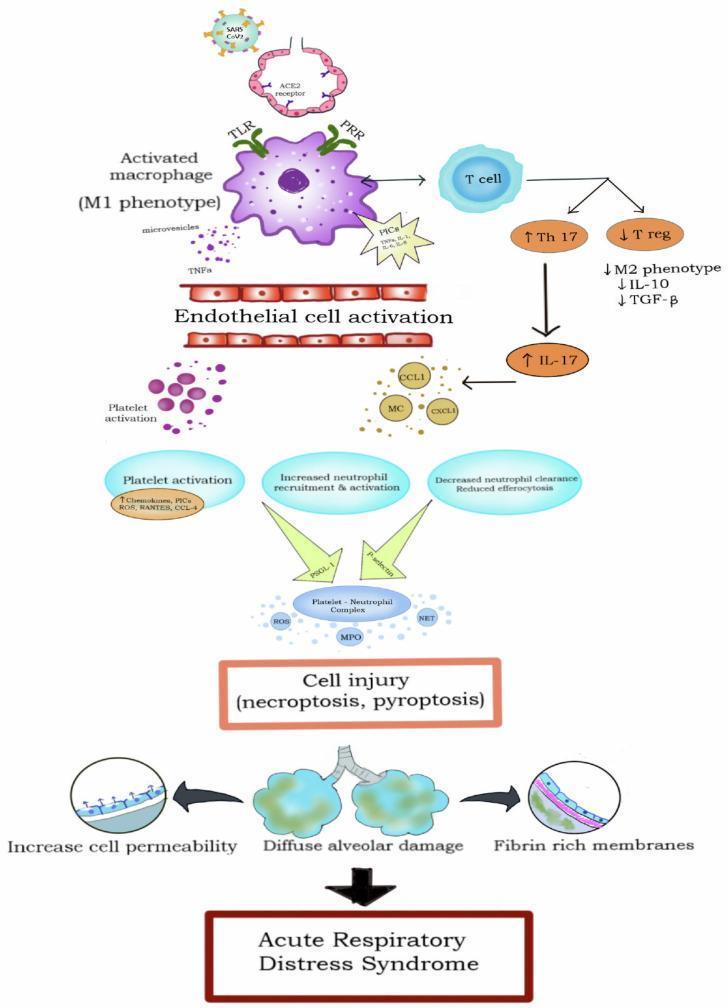
- Alveolar macrophages (M1 phenotype) activated via TLR/PRR pathways release proinflammatory cytokines: TNF-α, IL-1β, IL-6, IL-8
- This cytokine storm recruits and hyperactivates neutrophils, creating a feedforward loop
- T-cell dysregulation: Th17 responses increase (↑IL-17), while T-regulatory cells decrease, reducing anti-inflammatory mediators (IL-10, TGF-β)
- Endothelial cell activation by TNF-α and proinflammatory cytokines amplifies permeability
7. Surfactant Dysfunction
- Normal pulmonary surfactant (primarily phosphatidylcholine + surfactant proteins SP-A, SP-B, SP-C, SP-D) reduces alveolar surface tension and prevents collapse
- In ARDS, phospholipase A₂ (released during pancreatitis or by inflammatory cells) degrades surfactant
- Plasma proteins leaking into alveoli inhibit surfactant function
- Loss of surfactant → progressive alveolar collapse → reduced functional residual capacity (FRC) → intrapulmonary shunting and refractory hypoxemia
- There is a strong correlation between serum phospholipase A₂ levels and respiratory insufficiency in pancreatitis-associated ARDS — Murray & Nadel's
8. Coagulation-Inflammation Cross-Talk
Inflammation and coagulation are tightly coupled in ARDS:
- TNF-α → stimulates tissue factor expression on endothelial cells → activates extrinsic coagulation cascade → thrombin and fibrin formation
- TNF-α also inhibits fibrinolysis
- Fibrin fragments act as chemoattractants for neutrophils, amplifying inflammation
- Intravascular fibrin deposition causes microthrombi → pulmonary hypertension
- Angiopoietin-2 (released from activated endothelium) destabilizes the vessel wall and promotes permeability; angiopoietin-1 is protective — levels of Ang-2 are elevated in ARDS and correlate with severity
9. Sodium & Water Transport Failure
Under normal conditions, alveolar fluid is actively cleared by Na⁺/K⁺-ATPase and epithelial sodium channels (ENaC) on type II pneumocytes. In ARDS:
- Hypoxia and cytokine-mediated injury impair these transport mechanisms
- Failure of active sodium transport allows edema fluid to persist in airspaces even after capillary leak has been controlled
- Recovery of alveolar fluid clearance capacity is a key determinant of outcome
10. Ventilator-Induced Lung Injury (VILI)
ARDS creates a heterogeneous lung with aerated ("baby lung") and flooded/collapsed regions. Mechanical ventilation at high tidal volumes or pressures can:
- Overdistend the aerated lung (volutrauma/barotrauma)
- Promote cyclic atelectrauma in recruitable lung units
- Trigger biotrauma: mechanical stretch activates inflammatory signaling (NF-κB pathway), releasing additional cytokines that can spread injury systemically → multi-organ failure
This is why low tidal volume ventilation (6 mL/kg predicted body weight, plateau pressure ≤30 cmH₂O) is a cornerstone of ARDS management. — Goldman-Cecil Medicine
11. Pulmonary Hypertension in ARDS
Multiple mechanisms contribute:
- Hypoxic vasoconstriction (HPV): alveolar hypoxia constricts local vasculature
- Intravascular fibrin microthrombi obstruct vessels
- Compression by positive-pressure ventilation
- Mediator-driven vasoconstriction (thromboxane A₂, endothelin-1)
Summary Flowchart
Inciting Insult (sepsis, pneumonia, trauma, aspiration)
↓
Alveolar macrophage + pattern recognition receptor activation
↓
Cytokine storm (TNF-α, IL-1β, IL-6, IL-8)
↓
Neutrophil sequestration & activation in pulmonary capillaries
↓
ROS, NE, NETs, proteases released
↓
Endothelial & epithelial barrier disruption (cadherin degradation)
↓
Protein-rich exudate floods alveoli
+ Surfactant degradation / dysfunction
+ Coagulation activation → microthrombi
+ Sodium transport failure
↓
Diffuse Alveolar Damage (DAD)
- Hyaline membrane formation
- Alveolar collapse (↓FRC)
- Right-to-left shunting
- ↓ Compliance, refractory hypoxemia
↓
ARDS (± progression to fibrosis, pulmonary hypertension, multi-organ failure)
Key sources: Murray & Nadel's Textbook of Respiratory Medicine, Chapters 134 (ARDS) and 126 (Pancreatitis-associated lung injury); Goldman-Cecil Medicine, Chapter 91 (Mechanical Ventilation).
This is a shared conversation. Sign in to Orris to start your own chat.