g6pd deficiency
Reading File
Finding Sources
Searching PubMed
"glucose-6-phosphate dehydrogenase deficiency"[MeSH Terms]
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
Reading File
Reading File
I now have comprehensive information from multiple authoritative sources. Let me compile the full answer.
Glucose-6-Phosphate Dehydrogenase (G6PD) Deficiency
Overview
G6PD deficiency is the most common disease-producing enzyme abnormality in humans, affecting more than 400 million people worldwide. It is inherited as an X-linked recessive trait, so it primarily affects males (hemizygous). Females can be carriers (heterozygous) and, depending on the degree of X-inactivation (lyonization), may have variable enzyme activity ranging from normal to severely reduced.
Highest prevalence occurs in people with ancestry from sub-Saharan Africa, the Mediterranean, Middle East, and Southeast Asia. This geographic overlap with malaria-endemic regions is not coincidental - G6PD-deficient erythrocytes offer partial protection against Plasmodium falciparum because the oxidant stress induced by the parasite lyses the RBC before the parasite can complete its lifecycle.
Pathophysiology
The core mechanism is a failure of oxidant defense in red blood cells.
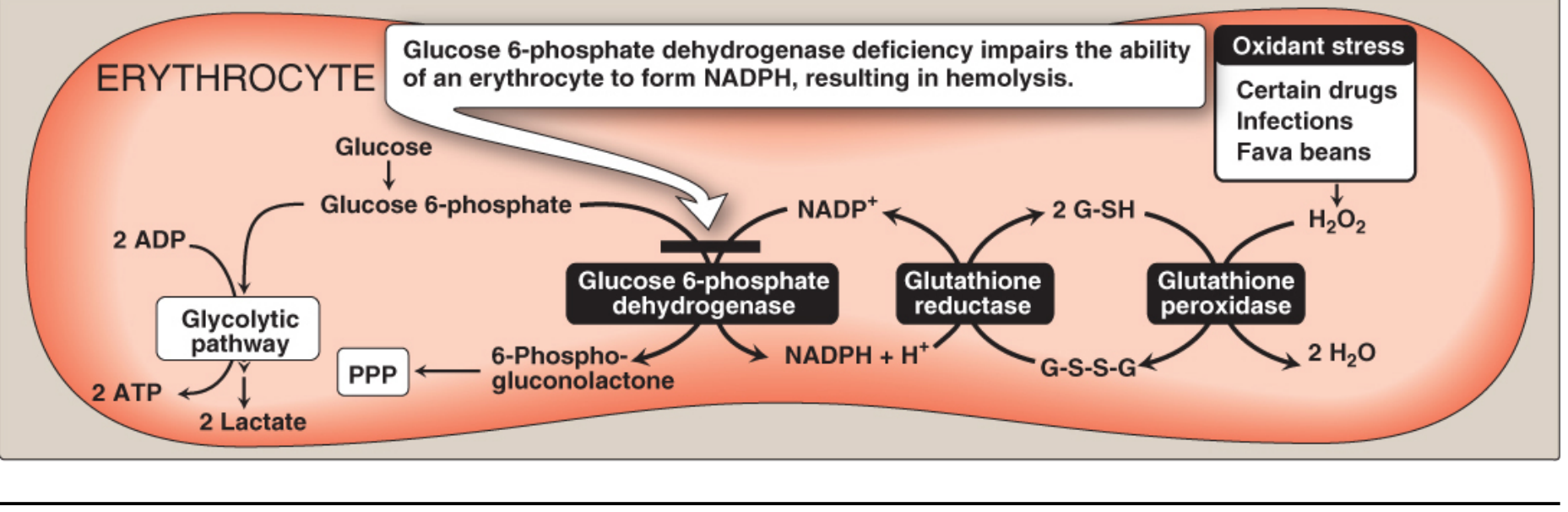
Why RBCs are uniquely vulnerable:
- The pentose phosphate pathway (PPP) is the only source of NADPH in red blood cells (other tissues can use the malic enzyme pathway).
- NADPH is required to regenerate reduced glutathione (G-SH) via glutathione reductase.
- G-SH neutralizes hydrogen peroxide (H₂O₂) via glutathione peroxidase.
- Without G6PD, NADPH fails, G-SH is depleted, H₂O₂ accumulates.
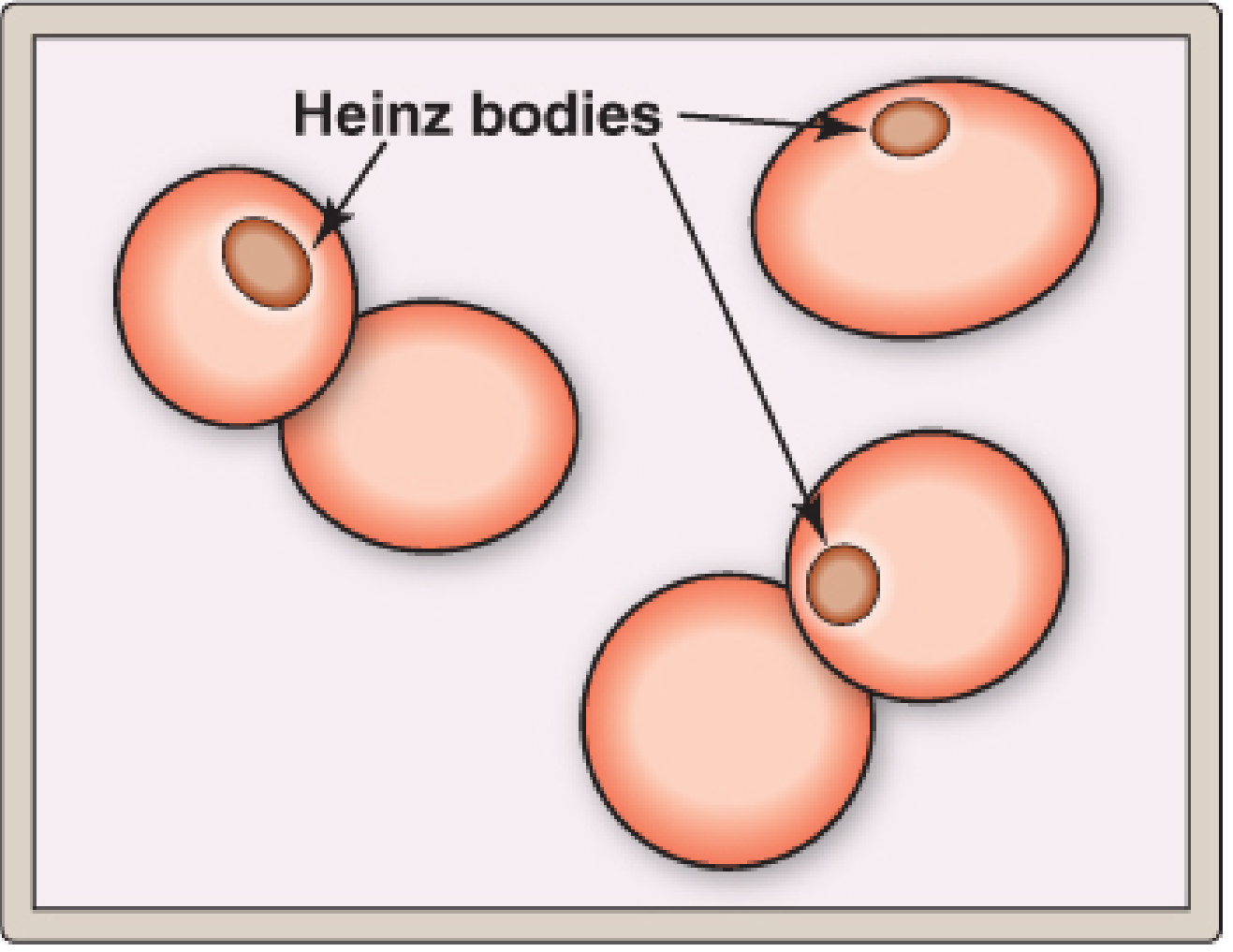
- Oxidized hemoglobin denatures into insoluble masses = Heinz bodies, which attach to the RBC membrane.
- Membrane rigidity increases, and the spleen removes these cells - producing hemolysis.
- RBCs also have no nucleus/ribosomes, so they cannot synthesize new G6PD enzyme. Older cells (with naturally declining enzyme) are most vulnerable.
Genetics and Variants
- The G6PD gene is located at Xq28. More than 400 variants have been identified; over 100 mutations described, mostly missense point mutations affecting enzyme kinetics, stability, or NADP+/G6P binding.
- Active G6PD is a homodimer or tetramer; mutations at subunit interfaces impair stability.
Key clinical variants:
| Variant | Population | Enzyme Activity | Half-life | Clinical Effect |
|---|---|---|---|---|
| GdB (normal) | 99% White Americans | Normal | ~60 days | None |
| GdA+ | 20% African Americans | Normal | Normal | None |
| GdA- | 10% African Americans | Mildly reduced | ~13 days | Intermittent hemolysis |
| GdMed | Mediterranean, India, SE Asia | Markedly reduced | Short | More severe hemolysis |
| GdCanton | Asian | Reduced | - | Similar to GdA- |
The GdA- variant has a shortened enzyme half-life (13 days vs. 60), so young RBCs have near-normal activity while older cells are severely deficient. This means after an acute hemolytic episode, as older cells are destroyed and replaced by young reticulocytes, the measured G6PD level may appear falsely normal - a diagnostic pitfall.
WHO Classification (5 Classes)
| Class | Enzyme Activity | Clinical Syndrome |
|---|---|---|
| I | Severely deficient | Congenital nonspherocytic hemolytic anemia (CNSHA) - spontaneous, no precipitant needed |
| II | Severely deficient (<10%) | Intermittent hemolysis (e.g., GdMed) |
| III | Mildly deficient (10-60%) | Hemolysis only after oxidant stress (e.g., GdA-) |
| IV | Normal (60-150%) | Asymptomatic |
| V | Elevated activity | Asymptomatic |
Classes II and III together account for >90% of G6PD variants.
Precipitating Factors (Triggers)
Affected individuals remain asymptomatic unless exposed to oxidant stress:
1. Drugs (the "A" categories):
- Antimalarials: Primaquine, pamaquine, chloroquine
- Sulfonamides/Sulfones: Dapsone, sulfamethoxazole-TMP, sulfanilamide
- Antibiotics: Nitrofurantoin, chloramphenicol, nalidixic acid
- Analgesics: Phenacetin (aspirin in moderate doses is generally acceptable; paracetamol is safe)
- Miscellaneous: Naphthalene (moth balls), methylene blue, vitamin K analogues, rasburicase
2. Favism - ingestion of fava (broad) beans, especially in the Mediterranean variant. Not all G6PD-deficient individuals develop favism, but all patients with favism have G6PD deficiency.
3. Infection - the most common precipitant. Macrophage-generated free radicals during the inflammatory response diffuse into RBCs and cause oxidative damage.
Clinical Manifestations
1. Acute Hemolytic Anemia (AHA)
Most dramatic presentation. Onset within 24-48 hours of oxidant exposure.
- Symptoms: Irritability, fever, nausea, abdominal pain, diarrhea
- Signs: Jaundice, hemoglobinuria (dark/"cola-colored" urine), pallor, hepatosplenomegaly
- Labs: Normochromic normocytic anemia, reticulocytosis, anisocytosis, poikilocytes, bite cells (splenic pitting of Heinz bodies), raised bilirubin, raised LDH, reduced haptoglobin
- Severe anemia may precipitate heart failure; acute renal failure may occur
2. Neonatal Jaundice (NNJ)
- Seldom present at birth; peak onset days 2-3 of life
- Severity variable; can be severe enough to cause kernicterus or death
- Anemia is rarely severe (unlike AHA)
- Risk increased in infants who also carry UGT1A1 polymorphism (Gilbert syndrome)
- Management same as NNJ from other causes - usually phototherapy
3. Chronic Nonspherocytic Hemolytic Anemia (CNSHA) - Class I
- Rare; caused by uncommon variants with severely impaired basal NADPH production
- Lifelong hemolysis without any external precipitant
- Severity varies - some compensated, others require intermittent or regular transfusions
Diagnosis
Screening:
- Fluorescence spot test: G6PD converts NADP+ → NADPH, which fluoresces under long-wave UV light. Rapid, inexpensive, high sensitivity. Semiquantitative only. Unreliable after acute hemolysis (young reticulocytes have normal activity) and may miss female heterozygotes.
Confirmatory:
- Spectrophotometric assay: Direct measurement of NADPH production. More sensitive but still requires ~20-30% G6PD-deficient cells for an abnormal result.
- DNA/molecular testing: Best for female heterozygotes and when hemolytic episode has just occurred.
- Timing matters: Test at least 2-3 months after an acute episode to avoid false-negative results.
Management
Acute Hemolytic Anemia:
- Remove the trigger - stop offending drug, treat underlying infection
- Most cases require no specific treatment beyond supportive care and hydration
- Severe anemia: blood transfusion (can be a medical emergency in children)
- Acute renal failure: hemodialysis if needed; recovery is the rule without pre-existing kidney disease
Chronic CNSHA:
- Regular folic acid supplementation
- Avoid hemolytic drugs and oxidant exposures
- Blood transfusions during exacerbations (usually triggered by intercurrent infection)
- Iron chelation if regular transfusions are required
- Splenectomy: no evidence of selective splenic destruction unlike hereditary spherocytosis, but has shown benefit in severe cases in practice
Neonatal jaundice: Standard management - phototherapy; exchange transfusion for severe hyperbilirubinemia
Prevention:
- Screening programs in high-prevalence populations
- Patient education: avoid triggers, carry a drug list
- Test before prescribing primaquine, dapsone, or other high-risk drugs
Sources:
- Lippincott Illustrated Reviews: Biochemistry, 8e, pp. 445-448 (G6PD Deficiency and Precipitating Factors)
- Goldman-Cecil Medicine, Ch. 147 (G6PD Clinical Manifestations, Diagnosis, Drug Table)
- Harrison's Principles of Internal Medicine, 22e (Management of AHA and CNSHA)
This is a shared conversation. Sign in to Orris to start your own chat.