Fibrous dysplasia oral pathology according to shafer's
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Loading Image
Finding Sources
Reading File
Reading File
I now have comprehensive content. Let me compile a detailed, Shafer's-style oral pathology answer on fibrous dysplasia based on the rich content gathered from these authoritative textbooks.
Fibrous Dysplasia - Oral Pathology (per Shafer's framework)
Note: Shafer's Textbook of Oral Pathology is not directly available in this library, but the content below is compiled from Robbins & Kumar Basic Pathology, Robbins Cotran & Kumar Pathologic Basis of Disease, Harrison's Principles of Internal Medicine, Cummings Otolaryngology, and Scott-Brown's Otorhinolaryngology - all of which align with Shafer's descriptions of this entity.
Definition
Fibrous dysplasia (FD) is a benign, chronic, slowly progressive bone disorder resulting from a localized developmental arrest, in which normal bone fails to differentiate into mature structures. Normal cancellous bone is progressively replaced by abnormal fibrous tissue containing poorly formed woven bone trabeculae.
Etiopathogenesis / Molecular Basis
All forms of fibrous dysplasia arise from somatic gain-of-function (activating) mutations in the GNAS1 gene on chromosome 20q13, which encodes the alpha subunit of the stimulatory G-protein (Gsα).
- The mutation constitutively activates Gsα, elevating cyclic AMP (cAMP) levels via the protein kinase A signal transduction pathway
- This promotes mesenchymal cell proliferation and disrupts osteoblast differentiation, causing fibroblast-like cells to produce imperfect woven bone instead of mature lamellar bone
- The phenotype depends on the stage of embryogenesis at which the postzygotic mutation occurs and which mesenchymal cells harbor it - this explains the mosaic (patchy) distribution of lesions
Classification
| Form | Description |
|---|---|
| Monostotic | Single bone involved; most common (~80%); diagnosed at 20-30 years; craniofacial involvement in ~25% of cases, with maxilla most commonly affected |
| Polyostotic | Multiple bones; typically manifests in children <10 years; maxilla and craniofacial bones frequently involved |
| McCune-Albright Syndrome (MAS) | Polyostotic FD + café-au-lait skin spots (rough "coast of Maine" borders) + endocrine hyperfunction (classically precocious puberty in girls) |
| Mazabraud Syndrome | Fibrous dysplasia + soft tissue myxoma |
Oral and Craniofacial Features
Sites of Involvement
- The maxilla is the most commonly affected jaw bone; the mandible is less frequently involved
- Craniofacial involvement occurs in up to 25% of monostotic cases and in >50% of polyostotic cases (skull involvement)
- Other craniofacial bones: frontal, sphenoid, temporal, orbital walls, ethmoid, and zygoma
- The temporal bone may be involved (mostly monostotic form, 70%)
Clinical Presentation
- Painless, slowly progressive facial swelling causing facial asymmetry
- Malocclusion due to jaw expansion
- Migration and mobility of teeth from jaw involvement
- Displacement or impaction of teeth
- In McCune-Albright - melanosis (café-au-lait pigmentation) of gingival tissues and facial skin
- Cranial nerve involvement - conductive hearing loss (most common, ~80% with temporal bone FD) from narrowing of the external auditory canal; rarely optic nerve compression, facial nerve palsy
- Proptosis from orbital involvement
- The disease starts early in life (childhood); the monostotic form tends to become quiescent at puberty, while polyostotic form may continue to progress
Radiographic Features
| Feature | Description |
|---|---|
| "Ground-glass" appearance | Classic finding - hazy, homogeneous opacity resembling ground glass, due to irregularly arranged spicules of woven bone in fibrous stroma |
| Radiolucent phase (lytic) | Well-defined, smooth or scalloped radiolucent area - early lesion |
| Radiopaque/fibrous phase | Areas of increased radiodensity as mineralization progresses |
| Expansion with cortical thinning | Lesion expands medullary cavity; periosteal reaction usually absent |
| Bowing deformities | Particularly in long bones; "shepherd's crook" deformity of femur in polyostotic disease |
The ground-glass appearance results from the mixture of fibrous tissue and irregularly oriented woven bone trabeculae, giving an overall opacity between pure bone and pure fibrous tissue.
Histopathology (Microscopic Features)
The characteristic histology consists of:
- Replacement of normal cancellous bone by a cellular fibroblastic stroma arranged in a whorled pattern
- Irregular curvilinear trabeculae of woven bone - often described as resembling "Chinese letters" or "alphabet soup"
- No rimming osteoblasts around the woven bone trabeculae (key distinguishing feature from ossifying fibroma, which has osteoblastic rimming)
- The stroma is moderately cellular fibroblastic proliferation without significant atypia
- Additional findings: cystic degeneration, hemorrhage, foamy macrophages, osteoclast-type giant cells
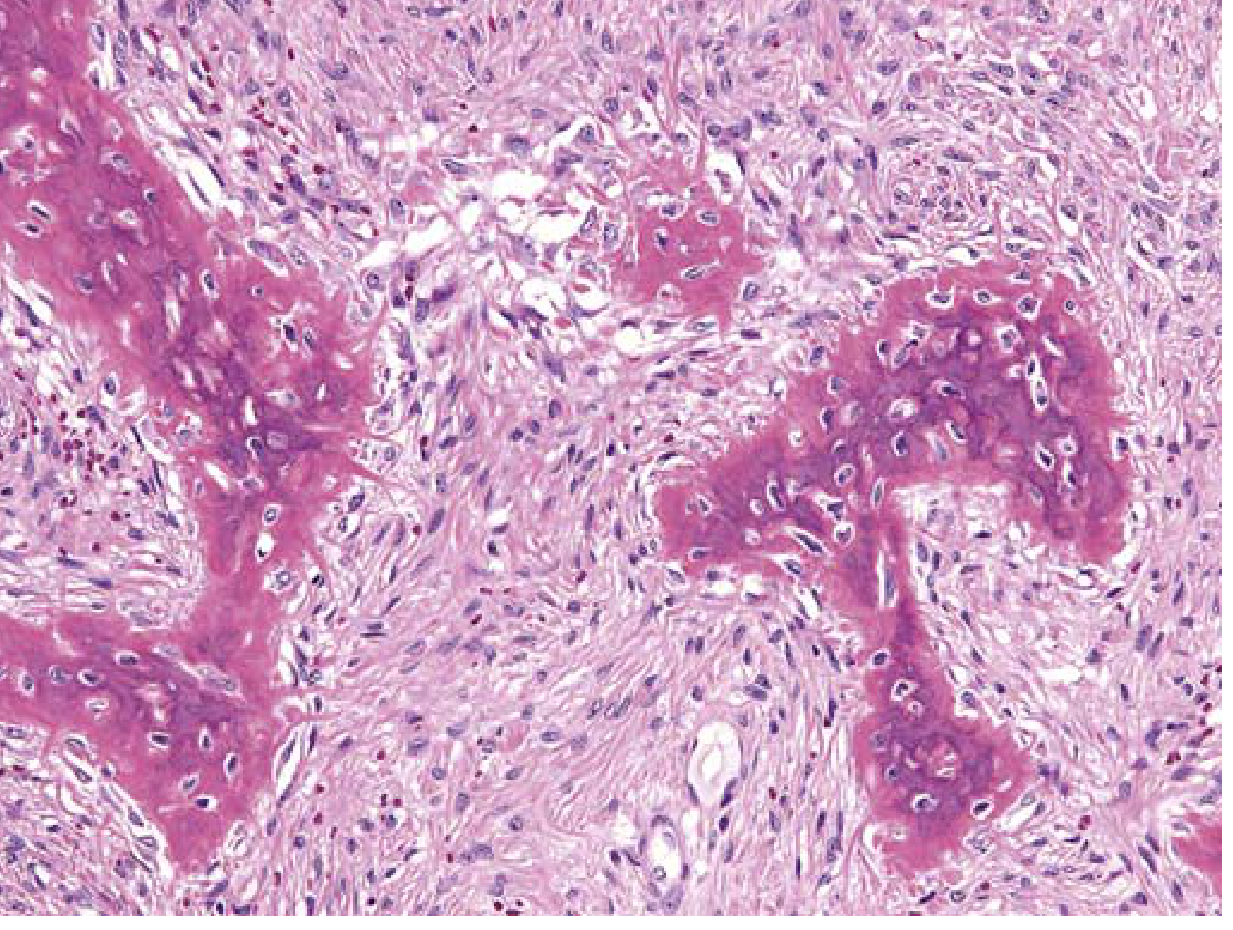
Fibrous dysplasia: Curvilinear trabeculae of woven bone surrounded by a moderately cellular fibroblastic proliferation. Note the absence of osteoblastic rimming around trabeculae.
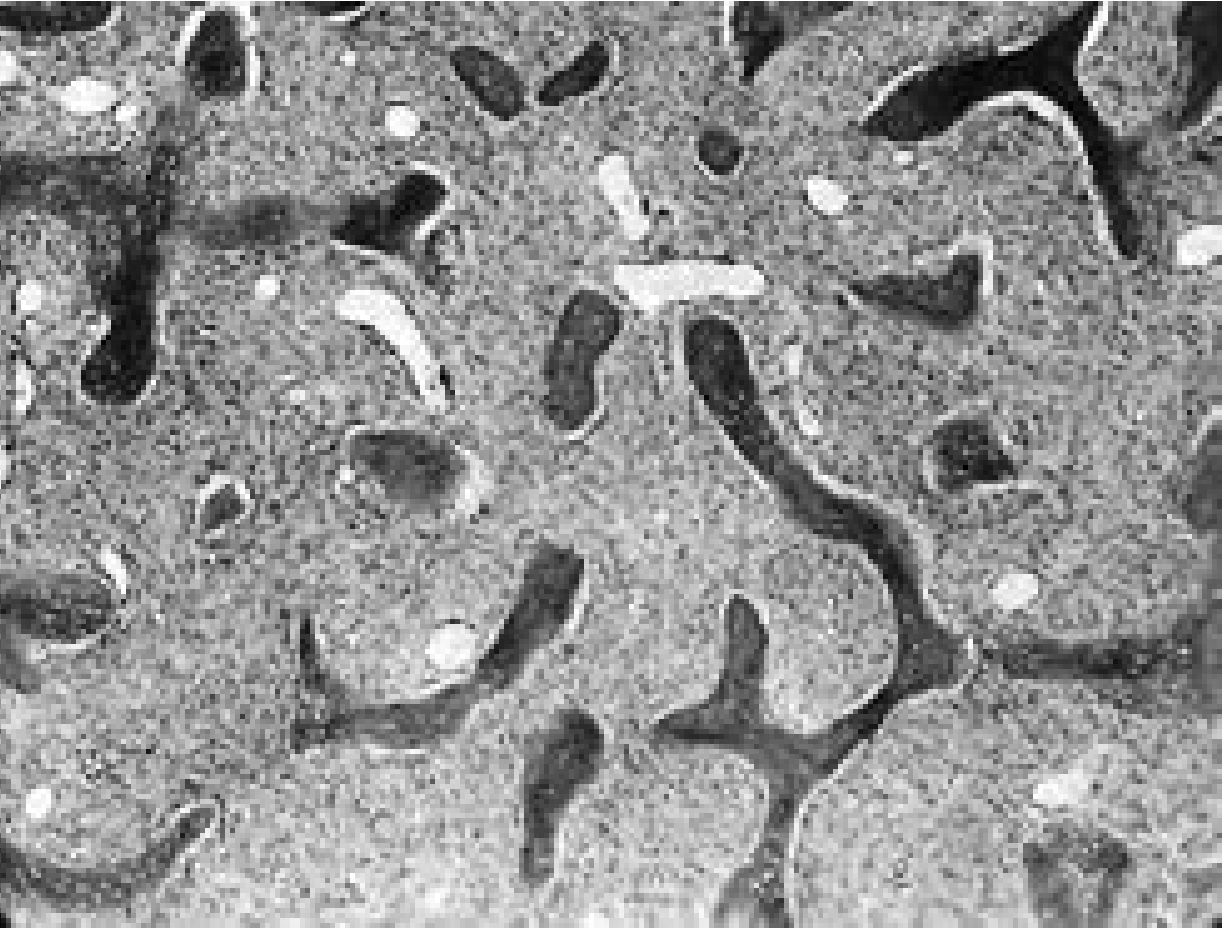
Fibrous dysplasia: Irregularly arranged spicules of woven bone in a fibrovascular stroma showing the characteristic whorled pattern (×64).
Laboratory Findings
- Serum calcium and phosphorus levels - usually normal
- Serum alkaline phosphatase - elevated in ~30% of polyostotic cases
- In rare widespread polyostotic cases: renal phosphate wasting, hypophosphatemia (due to a phosphaturic factor produced by abnormal fibrous tissue), which may cause rickets or osteomalacia
Differential Diagnosis
| Condition | Distinguishing Feature |
|---|---|
| Ossifying fibroma | Histologically: has osteoblastic rimming; odontogenic; well-encapsulated; affects mandible/maxilla tooth-bearing bone |
| Central giant cell granuloma | Giant cells prominent; younger patients; mandible more common |
| Cherubism | Hereditary (autosomal dominant); bilateral; involves mandible/maxilla; mandibular condyles spared |
| Paget's disease | Older patients; mosaic "jigsaw puzzle" pattern; elevated ALP; serum Ca/PO4 may be abnormal |
| Osteosarcoma | Malignant; rapid growth; periosteal reaction (Codman's triangle, sunburst) |
Complications
- Pathologic fractures - particularly in polyostotic/long bone disease
- Cranial nerve palsies - optic nerve compression, facial nerve palsy, SNHL
- Sarcomatous transformation - rare but serious; incidence ~0.4-1%; risk increased by radiation therapy; most commonly osteosarcoma or fibrosarcoma; involves facial bones or femur
- Recurrence after curettage - common
Treatment
- Conservative/watchful waiting - for small, asymptomatic, monostotic lesions
- Bisphosphonate therapy (e.g., pamidronate) - reduces severity of bone pain in polyostotic disease
- Surgical correction - for deformities, pathologic fractures, nerve decompression
- Curettage - for symptomatic lesions (recurrence is common)
- Surgical timing for craniofacial lesions: ideally after puberty when lesions tend to stabilize
- Radiotherapy is absolutely contraindicated - increases risk of malignant transformation
Key Points for Exam (Shafer's Highlights)
- Fibrous dysplasia = woven bone trabeculae WITHOUT osteoblastic rimming (vs. ossifying fibroma = WITH rimming)
- Mutation: GNAS1 gene → Gsα activation → elevated cAMP
- Radiograph: "ground-glass" appearance is pathognomonic
- Maxilla > mandible in jaw involvement
- McCune-Albright triad: polyostotic FD + café-au-lait spots ("coast of Maine") + endocrine dysfunction (precocious puberty in girls)
- Malignant transformation rare but possible; never irradiate as it increases this risk
- Histology classic: "Chinese letter" trabecular pattern in cellular fibrous stroma
Content synthesized from: Robbins & Kumar Basic Pathology (9780323790185); Robbins, Cotran & Kumar Pathologic Basis of Disease (9780443264528); Harrison's Principles of Internal Medicine 22E (9781265977061); Cummings Otolaryngology Head and Neck Surgery (9780323612173); Scott-Brown's Otorhinolaryngology Head & Neck Surgery Vol 1 (9781138094611)
This is a shared conversation. Sign in to Orris to start your own chat.