Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome" AND pathophysiology
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Loading Image
Loading Image
I now have all the content I need for a thorough, well-illustrated answer. Let me compile the complete response.
Mechanism of Acute Respiratory Distress Syndrome (ARDS)
Definition and Overview
ARDS is a clinical syndrome of respiratory insufficiency caused by diffuse alveolar damage (DAD) - the pathologic correlate of the disease. Unlike cardiogenic pulmonary edema (which results from elevated left-sided cardiac pressures causing hydrostatic fluid transudation), ARDS produces non-cardiogenic pulmonary edema: the alveolar-capillary barrier exhibits increased permeability, allowing protein-rich exudative fluid to flood the airspaces. The result is alveolar filling, shunting, decreased compliance, and life-threatening hypoxemia.
The Berlin definition requires: acute onset within 1 week of a known clinical insult, bilateral opacities on chest imaging not fully explained by effusions/collapse/nodules, PaO2/FiO2 ratio <300 mmHg with PEEP ≥5 cmH2O, and respiratory failure not fully explained by cardiac failure.
Triggers
ARDS arises from direct pulmonary insults or systemic conditions:
| Direct (pulmonary) | Indirect (extrapulmonary) |
|---|---|
| Pneumonia (bacterial, viral, fungal) | Sepsis (most common overall trigger) |
| Aspiration of gastric contents | Severe trauma/polytrauma |
| Pulmonary contusion | Pancreatitis |
| Inhalation injury / near-drowning | Massive transfusion (TRALI) |
| Diffuse alveolar hemorrhage | Burns, drug overdose |
Three Sequential Phases
The time course of ARDS divides into three overlapping phases:

ARDS phase timeline - from exudative (edema, hyaline membranes), through proliferative (interstitial inflammation), to fibrotic (structural remodeling). Reproduced from Harrison's Principles of Internal Medicine, 22E.
Phase 1 - Exudative Phase (Days 0-7)
This is the acute inflammatory phase that produces the classic picture of ARDS.
Step 1: Initiating Injury and Pattern Recognition
The trigger - whether sepsis-derived LPS, aspiration, a virus, or trauma-associated DAMPs - activates Toll-like receptors (TLRs) on resident alveolar macrophages and type I alveolar epithelial cells (ATI cells). This drives secretion of pro-inflammatory chemokines (IL-8/CXCL8, MCP-1) and cytokines (TNF-α, IL-1β, IL-6) into the alveolar space and systemic circulation.
Step 2: Neutrophil Sequestration and Transmigration
One of the earliest signs - occurring even before hypoxemia - is transient leukopenia due to neutrophil sequestration in the pulmonary microvasculature. The pulmonary capillary lumen (average 5 µm) is narrower than the average neutrophil diameter (~8 µm). Normally, neutrophils deform to pass through. When activated by chemokines or LPS, neutrophils undergo cytoskeletal actin polymerization that makes them "stiff," trapping them in pulmonary capillary segments. Once sequestered, neutrophils do not require the usual selectin/integrin adhesion molecules to migrate further into the interstitium. - Murray & Nadel's Textbook of Respiratory Medicine, p. 3147
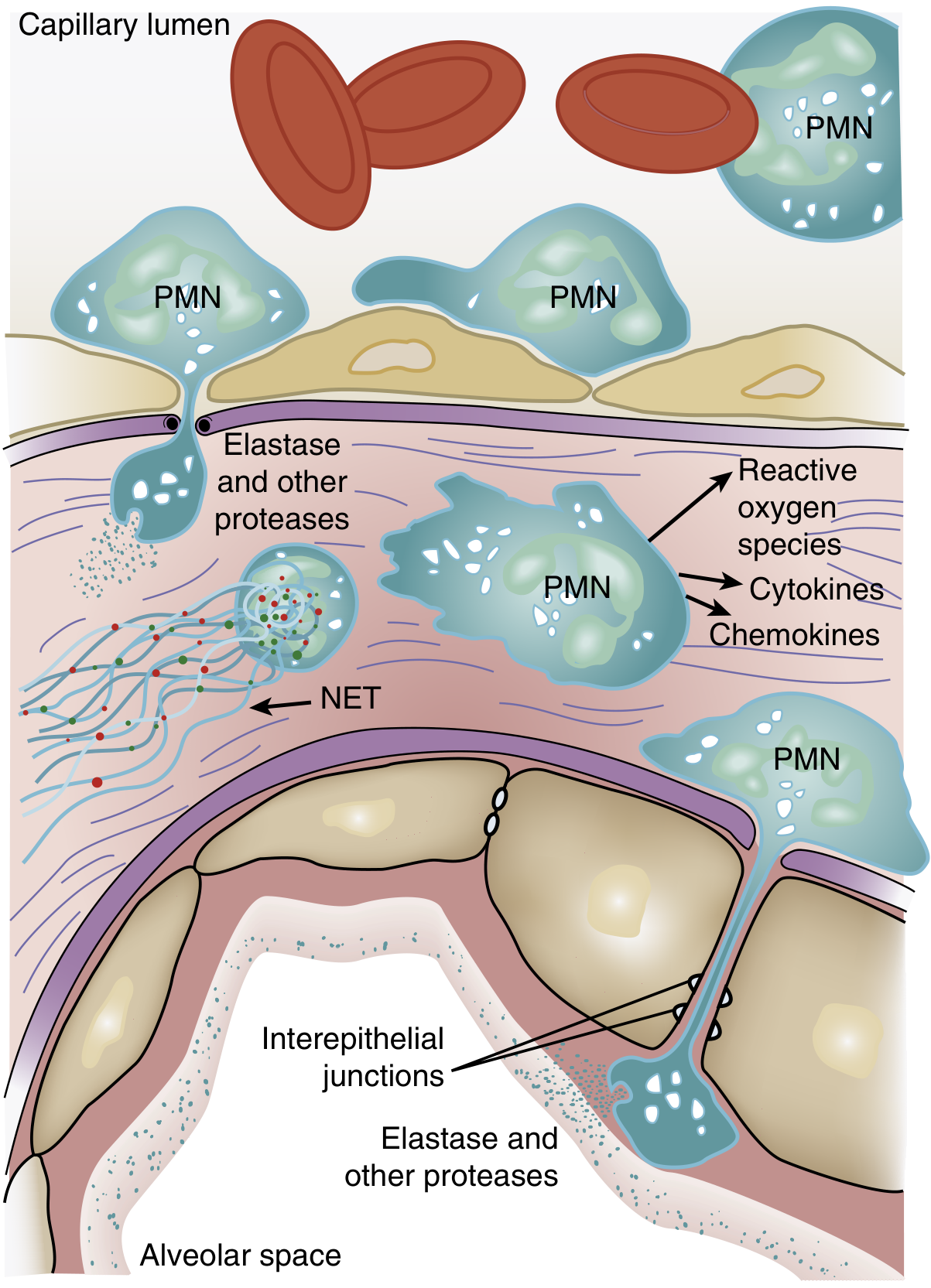
PMNs (neutrophils) exit the capillary lumen, transmigrate across the alveolar-capillary membrane, and release elastase, other proteases, reactive oxygen species (ROS), cytokines, chemokines, and neutrophil extracellular traps (NETs) into the interstitium and alveolar space. Reproduced from Murray & Nadel's Textbook of Respiratory Medicine.
Step 3: Alveolar-Capillary Barrier Disruption
The sequestered and transmigrating neutrophils release a destructive arsenal:
- Proteases (neutrophil elastase, matrix metalloproteinases): degrade structural proteins of the extracellular matrix and tight junction proteins (e.g., occludin, ZO-1), dismantling the physical barrier
- Reactive oxygen species (ROS): oxidize lipids and proteins in cell membranes, causing direct epithelial and endothelial cell death via necrosis and apoptosis
- Neutrophil extracellular traps (NETs): web-like chromatin-histone-enzyme structures that contribute to both pathogen killing and host tissue injury
- Platelet-PMN aggregates: activated platelets form aggregates with neutrophils and monocytes, amplifying the inflammatory signal and promoting microvascular occlusion
Monocyte-derived macrophages also migrate in and can trigger epithelial cell apoptosis via IFN-β-dependent release of TRAIL (TNF-related apoptosis-inducing ligand). - Harrison's Principles of Internal Medicine, 22E, p. 2343-2344
The net result of this cellular assault is a two-front barrier failure:
- Microvascular endothelial barrier breakdown - necessary and sufficient to produce ARDS edema
- Alveolar epithelial barrier breakdown - especially type I pneumocyte (ATI) destruction
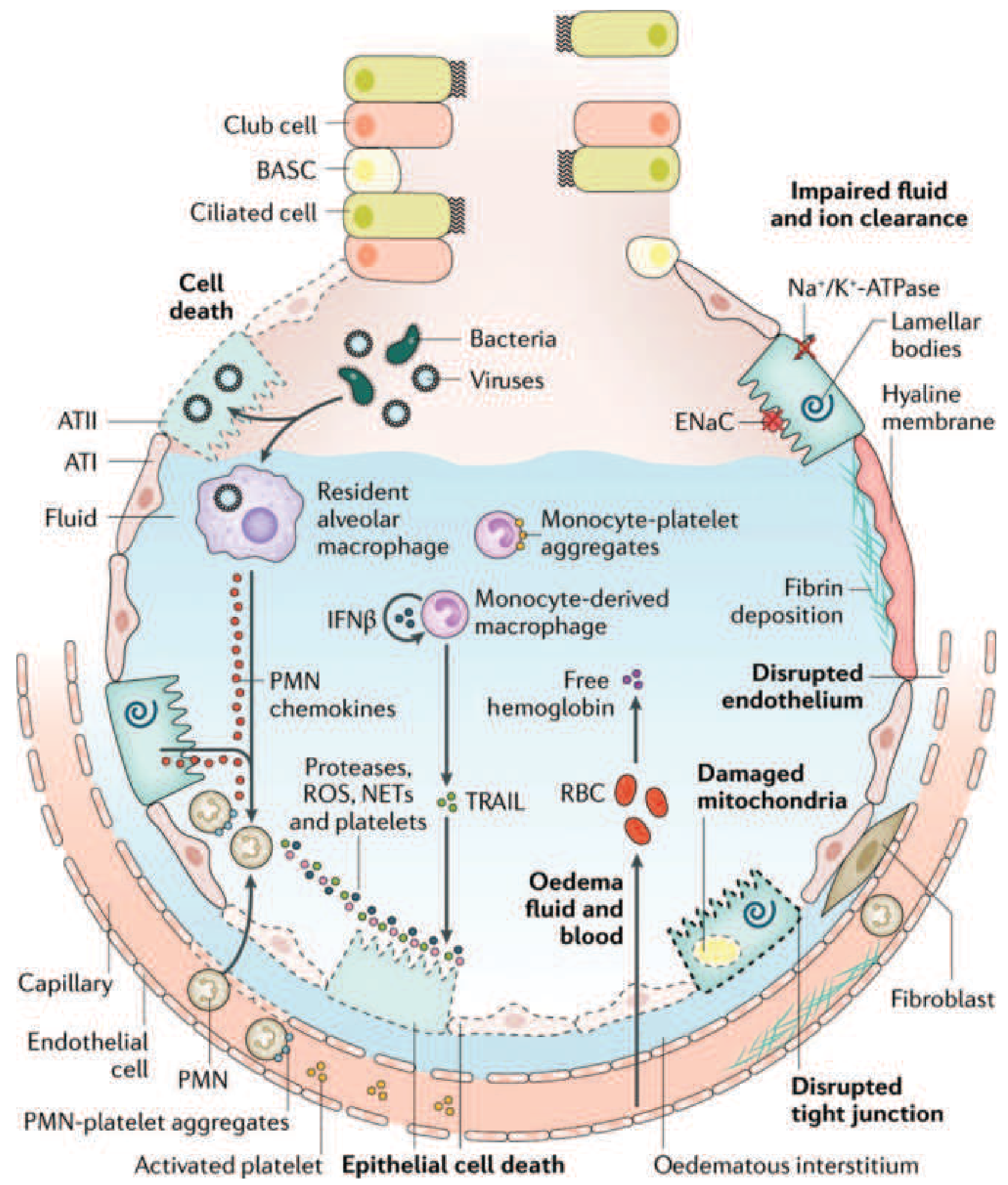
Comprehensive diagram of the injured alveolus in the acute (exudative) phase of ARDS: neutrophil-mediated injury pathways, monocyte recruitment, platelet activation, RBC extravasation releasing free hemoglobin, disrupted tight junctions, fibrin deposition, impaired Na+/K+-ATPase-driven fluid clearance, and hyaline membrane formation. Reproduced from Harrison's Principles of Internal Medicine, 22E (adapted from Matthay MA et al, Nat Rev Dis Primers 2019).
Step 4: Alveolar Flooding and Edema Formation
With both barriers breached, protein-rich exudative fluid pours from the capillary lumen into the interstitium and alveolar space. This is distinct from the low-protein transudative edema of heart failure. The edema fluid contains plasma proteins, fibrin, inflammatory cells, and red blood cells.
Simultaneously:
- Impaired alveolar fluid clearance: Normally, type II pneumocytes drive Na+ transport via epithelial sodium channels (ENaC) with the Na+/K+-ATPase, creating osmotic gradient to clear alveolar fluid. Injury and hypoxemia impair this pump, so edema cannot be cleared efficiently
- The edema distributes unevenly - predominantly in gravity-dependent lung zones - creating a heterogeneous injury pattern visible on CT
Step 5: Surfactant Depletion and Dysfunction
Type II pneumocytes (ATII cells) produce surfactant; in ARDS they are injured, leading to:
- Decreased surfactant production of its active phospholipid components (dipalmitoylphosphatidylcholine, phosphatidylglycerol)
- Reduced ratio of large (active) to small (inactive) surfactant aggregates
- Plasma proteins that leak into alveolar space inhibit surfactant function
- Neutrophil elastase directly degrades surfactant protein A (SP-A)
Loss of surfactant increases alveolar surface tension, promoting alveolar collapse (atelectasis) and further reducing lung compliance. - Murray & Nadel's Textbook of Respiratory Medicine, p. 3146
Step 6: Hyaline Membrane Formation
Condensed plasma proteins, fibrin, cellular debris, and dysfunctional surfactant components aggregate in the airspaces to form hyaline membrane whorls - the pathologic hallmark of DAD. These line the denuded alveolar walls and block gas exchange.
Step 7: Pulmonary Vascular Injury and Hypertension
Vascular changes occur early:
- Microthrombus formation: activated platelets and coagulation cascade generate fibrin-platelet thrombi in small pulmonary arterioles and capillaries
- Hypoxic vasoconstriction: alveolar hypoxia triggers vasoconstriction of local pulmonary arterioles (HPV), redistributing flow to ventilated areas - but when diffuse, this raises pulmonary vascular resistance throughout
- Compression by positive-pressure ventilation and intra-vascular fibrin deposition further increase pulmonary vascular resistance
The consequence is pulmonary hypertension and increased dead space (blood flow to non-ventilated regions creates intrapulmonary shunt; reduced flow to ventilated regions creates dead-space ventilation). This explains why ARDS patients often develop hypercapnia alongside hypoxemia.
Gas Exchange Failure - The Physiologic Consequence
The combined effect produces severe hypoxemia that is refractory to supplemental oxygen alone (hallmark of ARDS), because the shunt bypasses alveolar gas entirely. Lung compliance falls due to edema and atelectasis, so the work of breathing increases dramatically, leading to dyspnea, tachypnea, and ultimately respiratory fatigue requiring mechanical ventilation.
Phase 2 - Proliferative Phase (Days 7-21)
If the patient survives the exudative phase, the proliferative phase involves both repair and potential fibroproliferation:
- Type II pneumocyte hyperplasia: ATII cells proliferate to resurface the denuded alveolar epithelium (they are the progenitor cells for type I pneumocytes). The alveolar surface becomes cuboidal in appearance
- Fibroblast influx: activated fibroblasts migrate in and begin depositing interstitial and intra-alveolar collagen. Importantly, elevated N-terminal procollagen peptide III in BAL fluid can be detected as early as 24 hours after ARDS onset, meaning fibroproliferation may start simultaneously with the initial inflammatory injury - not just after it
- Edema reabsorption: partial resorption of edema fluid characterizes this phase
- Fibrin organization: fibrin becomes prominent in alveoli and interstitium, thickening the air-blood barrier
- Neutrophil density decreases; the inflammatory milieu shifts toward resolution or - in some patients - progressive fibrosis
Many patients recover during this phase. Some, however, develop progressive lung injury and early pulmonary fibrosis. - Fishman's Pulmonary Diseases and Disorders, p. 2480
Phase 3 - Fibrotic Phase (>Day 21)
Not all patients enter this phase - it is linked to prolonged mechanical ventilation and severe initial injury. Features include:
- Alveolar septal thickening from organizing fibrosis
- Bullae formation (air cysts from tissue destruction and remodeling)
- Loss of pulmonary capillary bed: obliteration of pulmonary capillaries, reduced vascular surface area
- Structural distortion: architectural remodeling that may be permanent
Patients remaining in this phase have severely reduced lung compliance and persistent hypoxemia, and may require prolonged ventilatory support.
The CT scans below show this progression:
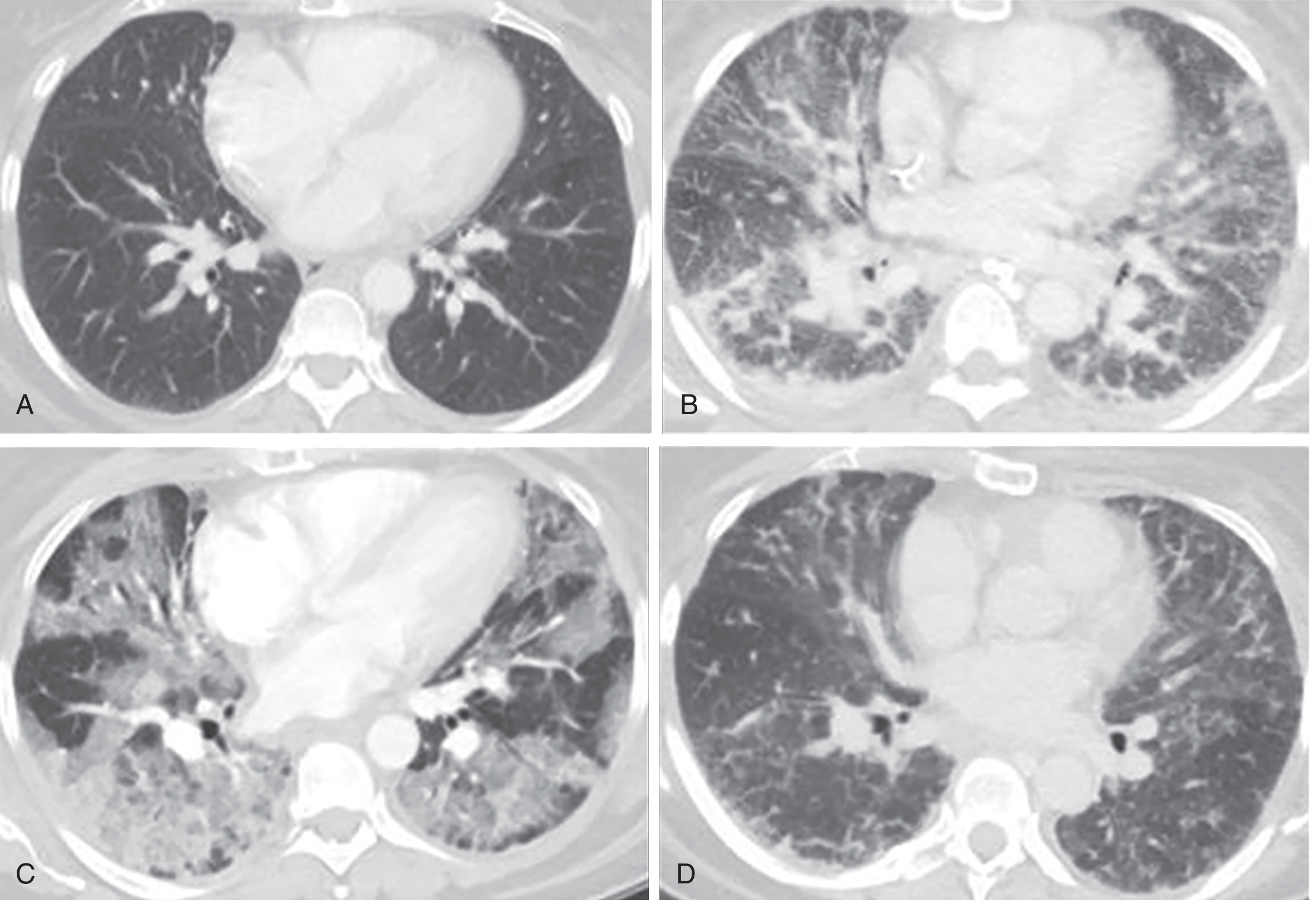
CT correlation of ARDS phases: (A) Normal baseline. (B) Exudative phase - bilateral ground-glass opacities and reticular changes without pleural effusions. (C) Proliferative phase - architectural distortion and dependent consolidation. (D) Fibrotic phase - linear and reticular abnormalities, particularly anterior. Reproduced from Murray & Nadel's Textbook of Respiratory Medicine.
Mechanistic Summary
TRIGGER (sepsis, pneumonia, trauma, aspiration)
↓
TLR activation on alveolar macrophages + ATI cells
↓
Cytokine/chemokine storm: TNF-α, IL-1β, IL-6, IL-8, LTB4
↓
Neutrophil sequestration in pulmonary microvasculature
↓
Neutrophil transmigration: ROS + proteases + NETs + cytokines
↓
Endothelial + epithelial (ATI) barrier disruption
↓
┌─ Protein-rich alveolar flooding (exudative edema)
├─ Surfactant depletion → alveolar collapse
├─ Hyaline membrane formation (DAD)
├─ Microvascular thrombosis → dead-space ↑
└─ Impaired Na+/K+ fluid clearance
↓
Hypoxemia (shunt) + Hypercapnia (dead space)
+ Reduced compliance + Pulmonary hypertension
↓ (days 7-21)
ATII hyperplasia, fibroblast activation, collagen deposition
↓ (>21 days, in subset)
Progressive fibrosis, architectural destruction
Key Points on DAD vs. ARDS
It is worth noting that only approximately 50% of ARDS patients show pathologic DAD on autopsy or biopsy. Those with DAD tend to be younger, more severely ill, have lower PaO2/FiO2 ratios, poorer compliance, and a 5-fold higher likelihood of death from hypoxemic respiratory failure. The other 50% have alternative pathologies (pneumonia, diffuse alveolar hemorrhage, eosinophilic pneumonia) that clinically mimic ARDS - which has important implications for targeted therapy. - Murray & Nadel's Textbook of Respiratory Medicine, p. 3146
Sources:
- Murray & Nadel's Textbook of Respiratory Medicine, Chapter 134 (Etiology and Pathogenesis of ARDS)
- Harrison's Principles of Internal Medicine, 22E, Chapter 312 (Clinical Course and Pathophysiology)
- Fishman's Pulmonary Diseases and Disorders, Chapter 140 (Pathologic Findings)
- Robbins & Kumar Basic Pathology, Chapter 11 (Acute Lung Injury and ARDS)
- Goldman-Cecil Medicine, Chapter 90 / 79 (Pathobiology)
This is a shared conversation. Sign in to Orris to start your own chat.