
20260715_165225.jpg
Explain
Reading File
Finding Sources
analyse_media
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Now I have all the detail I need. Here is a thorough explanation of every hallmark on that page.
Hallmarks of Cancer - Full Explanation
This is page 217 from First Aid for the USMLE Step 1, Pathology - Neoplasia section. Cancer is fundamentally driven by acquired DNA mutations that hijack normal cellular processes controlling growth, survival, and repair.
1. Growth Signal Self-Sufficiency
Normal cells only divide when told to by external growth factors. Cancer cells generate their own growth signals through mutations in:
| Gene Type | Example | How It Drives Cancer |
|---|---|---|
| Proto-oncogenes (encode growth factors) | PDGF in brain tumors | Tumor makes its own growth factor - autocrine loop |
| Growth factor receptors | HER2 (breast cancer) | Receptor fires constitutively even without ligand |
| Signaling molecules | RAS | Stuck in "ON" position, relaying constant grow signals |
| Transcription factors | MYC | Directly activates hundreds of genes promoting proliferation |
| Cell cycle regulators | Cyclins, CDKs | Bypass checkpoint controls; push through G1-S transition |
Key concept: Proto-oncogenes become oncogenes via gain-of-function mutations. Only ONE mutated allele is enough (dominant).
2. Anti-Growth Signal Insensitivity
Normal cells respond to "stop dividing" signals. Cancer cells ignore them via:
-
Tumor suppressor gene mutations (eg, Rb): The retinoblastoma protein (Rb) normally acts as a brake on the cell cycle. When Rb is phosphorylated and inactivated by cyclins/CDKs, the cell proceeds through G1. In cancer, Rb is lost/mutated, so the brakes are permanently off. Requires loss of BOTH alleles (recessive - "two-hit hypothesis").
-
Loss of E-cadherin / contact inhibition: E-cadherin is an adhesion molecule that tells a cell "I'm surrounded by neighbors, stop dividing." When lost, cells lose contact inhibition and pile up. The NF2 protein (merlin) acts downstream of E-cadherin to mediate this signal - NF2 mutations cause neural tumors (neurofibromatosis type 2). - Robbins Basic Pathology, p. 237
3. Evasion of Apoptosis
Apoptosis is programmed cell death - the body's failsafe for damaged or abnormal cells. Cancer cells circumvent this via two main mechanisms:
-
TP53 mutations: p53 is the "guardian of the genome." DNA damage normally activates p53 → cell cycle arrest (via p21) → DNA repair or apoptosis (via PUMA, BAX). When TP53 is mutated (found in >50% of all human cancers including lung, colon, breast), cells with damaged DNA survive and keep dividing. - Robbins Basic Pathology, p. 236-237
-
BCL2 overexpression: BCL2 is an anti-apoptotic protein. In follicular B cell lymphoma, the t(14;18) translocation fuses BCL2 to the immunoglobulin heavy chain gene, causing massive BCL2 overexpression. Lymphocytes accumulate because they can't die - the tumor grows slowly but relentlessly. - Robbins Basic Pathology, p. 145
4. Limitless Replicative Potential
Normal cells can divide only ~70 times before telomeres (protective caps on chromosome ends) shorten critically, triggering senescence or apoptosis.
The mechanism:
- Normal cells → telomeres shorten with each division → replicative senescence → cell death
- Cancer cells → reactivate telomerase (an enzyme that adds telomere sequences back) → telomeres are maintained → cells become immortal
Without telomerase reactivation, cancer cells would undergo "bridge-fusion-breakage" cycles from critically short telomeres, culminating in mitotic catastrophe and death. Telomerase reactivation rescues them from this fate. - Robbins Basic Pathology, p. 152-153
5. Sustained Angiogenesis
Tumors beyond ~1-2 mm need their own blood supply (diffusion alone is insufficient). They achieve this by tipping the balance toward pro-angiogenic signals:
- Increased VEGF (Vascular Endothelial Growth Factor) - produced by tumor or stromal cells
- Decreased inhibitors (eg, thrombospondin-1)
- Vessels sprout from existing capillaries (neoangiogenesis) or new endothelial cells are recruited from bone marrow (vasculogenesis)
- Tumor vessels are typically leaky and dilated (irregular architecture), which also facilitates tumor cell escape into the bloodstream
Clinical relevance: Anti-VEGF drugs (eg, bevacizumab) exploit this hallmark therapeutically.
6. Warburg Effect (Altered Cellular Metabolism)
Named after Otto Warburg (Nobel Prize 1931), this is the observation that cancer cells preferentially use aerobic glycolysis - converting glucose to lactate even when oxygen is plentiful. This seems inefficient (glycolysis yields 2 ATP vs. ~36 ATP from oxidative phosphorylation), but it provides a key advantage:
Why glycolysis? It rapidly generates the carbon building blocks (nucleotides, amino acids, lipids) that rapidly dividing cells need to build new cellular structures - not just energy. The byproduct is increased lactic acid production.
Clinical use: PET scans exploit this - 18F-fluorodeoxyglucose (FDG) is avidly taken up by metabolically hyperactive tumor cells, making tumors "light up" on PET. - Robbins Basic Pathology, p. 237-238
7. Immune Evasion
The immune system routinely identifies and destroys nascent cancer cells (immune surveillance). Successful tumors escape using multiple strategies:
| Mechanism | Explanation |
|---|---|
| Downregulate MHC class I | Cytotoxic T cells (CD8+) recognize tumor antigens presented on MHC-I. Less MHC-I = T cells can't see the tumor |
| Secrete TGF-β | Potent immunosuppressive cytokine; also recruits Regulatory T cells (Tregs) which dampen immune responses |
| Upregulate immune checkpoints | Eg, tumor cells express PD-L1, which binds PD-1 on T cells and turns them off ("T cell exhaustion") |
Clinical relevance: This hallmark is the basis of checkpoint inhibitor therapy (anti-PD-1, anti-CTLA-4), a major advance in cancer treatment.
8. Tissue Invasion
The step-by-step molecular process by which cancer cells breach the basement membrane and invade local tissue:
Loss of E-cadherin
→ Loosening of intercellular junctions
→ Metalloproteinases (MMPs) degrade basement membrane and ECM
→ Tumor cells attach to ECM proteins (laminin, fibronectin)
→ Cells migrate through degraded ECM ("locomotion")
→ Enter blood/lymphatic vessels (vascular dissemination)
Loss of E-cadherin also triggers Epithelial-to-Mesenchymal Transition (EMT) - cancer cells acquire a migratory, mesenchymal phenotype. TGF-β is a key driver of EMT. - Robbins Basic Pathology, p. 42
9. Metastasis
The spread of cancer cells from the primary tumor to distant organs. Steps:
- Local invasion (as above) → enter lymphatics or blood vessels
- Survival in circulation (most cells die here)
- Adhesion to endothelium at the target organ
- Extravasation (exit vessels) and homing to new tissue
- Colonization - grow in the new microenvironment
Predictable patterns:
- The target organ is often the first capillary bed encountered after leaving the primary tumor (eg, colon cancer → portal circulation → liver)
- Some cancers show organ tropism regardless of anatomy (eg, lung cancer metastasizes to adrenals; breast cancer to bone, brain, lung, liver)
Summary Mnemonic: "GEALW-IIM"
| Letter | Hallmark |
|---|---|
| G | Growth signal self-sufficiency |
| E | Evasion of apoptosis |
| A | Anti-growth signal insensitivity |
| L | Limitless replicative potential |
| W | Warburg effect |
| I | Immune evasion |
| I | Invasion |
| M | Metastasis |
These hallmarks are the conceptual backbone of oncology - every cancer drug target maps onto one or more of these mechanisms. Good luck with your USMLE prep!

20260715_172340.jpg
Explain
analyse_media
Finding Sources
analyse_media
Reading File
Reading File
Finding Sources
Loading Image
Reading File
I now have everything needed for a comprehensive explanation. Here it is:
Page 218 - Immune Checkpoint Interactions & Cancer Epidemiology
Part 1: Immune Checkpoint Interactions
Background - What is a Checkpoint?
T cells need two signals to become activated and attack a target:
- Signal 1: T cell receptor (TCR) recognizes antigen presented on MHC (on APC or tumor cell)
- Signal 2 (costimulation): CD28 on the T cell binds B7 on the APC → activating "+", proliferative signal
Without Signal 2, the T cell becomes anergic (unresponsive) - it "shuts off."
Immune checkpoints are molecular brakes that exploit this "Signal 2" requirement. Tumors hijack these brakes to hide from the immune system. Checkpoint inhibitor drugs block these brakes to re-unleash T cells against cancer.
Here is a diagram from Roitt's Essential Immunology showing how these checkpoints work and where the drugs target:
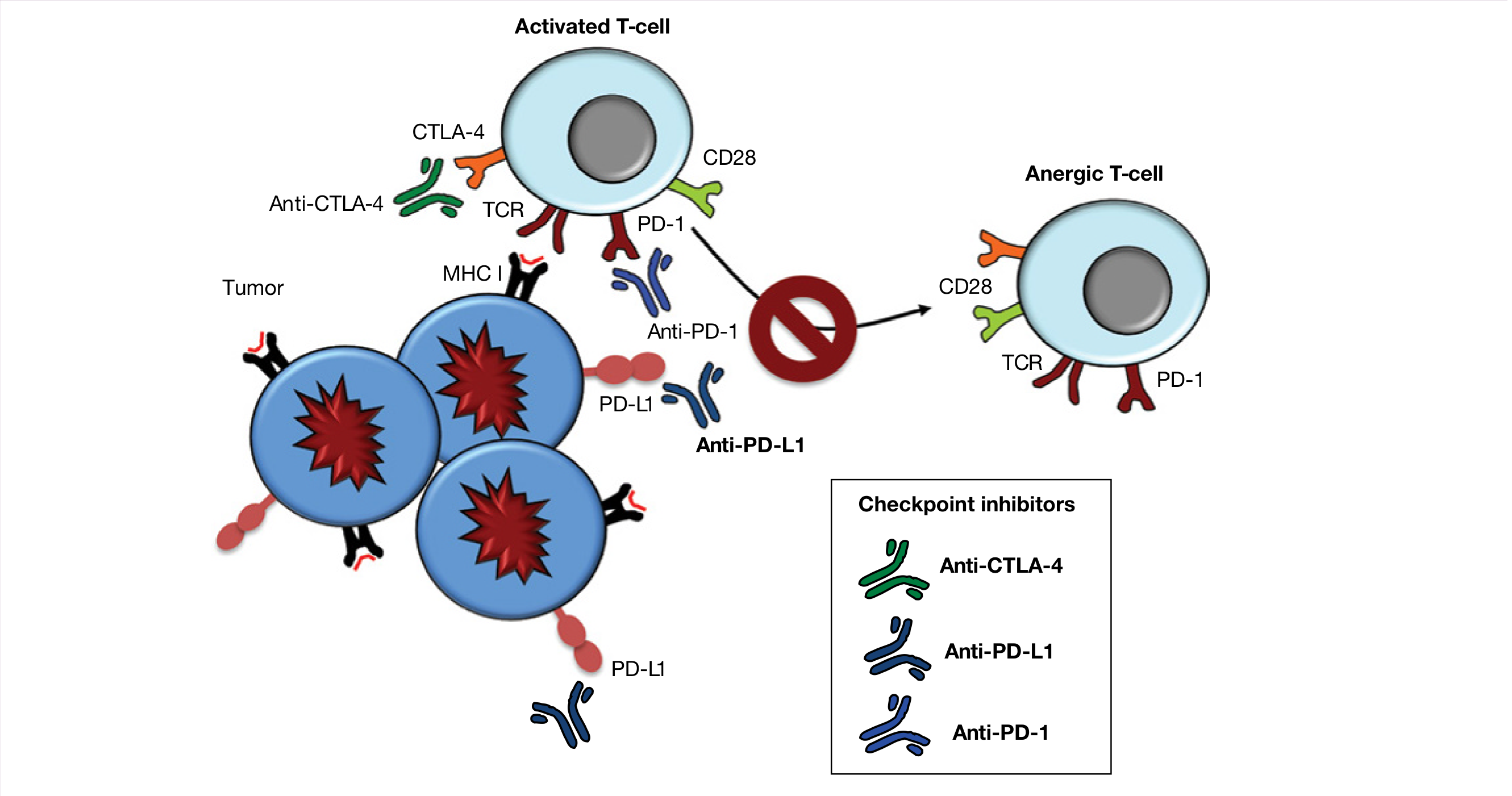
Checkpoint 1: CTLA-4 Pathway
The normal mechanism:
APC has B7 on its surface
├─ CD28 on T cell binds B7 → (+) ACTIVATION signal → T cell proliferates
└─ CTLA-4 (expressed LATER on activated T cells) also binds B7
but with HIGHER affinity than CD28
→ OUTCOMPETES CD28 for B7
→ Removes the costimulatory (+) signal
→ T cell activation is turned OFF
Why cancer exploits this: In the tumor microenvironment, CTLA-4 dominates and shuts down the anti-tumor T cell response before it gets going.
The drug - Ipilimumab (anti-CTLA-4):
- A monoclonal antibody that blocks CTLA-4
- CTLA-4 can no longer compete with CD28
- CD28 binds B7 freely → costimulatory signal restored → T cells attack tumor
- First approved in 2011 for advanced melanoma
- Side effects: autoimmunity - colitis, hepatitis, dermatitis (because you've removed a normal brake) - Cellular & Molecular Immunology, p. 971-973
Checkpoint 2: PD-1 / PD-L1 Pathway
The normal mechanism:
Tumor cell expresses PD-L1 on its surface
→ PD-L1 binds PD-1 on activated T cells
→ Activates ITIM motif → phosphatases dampen TCR signaling
→ T cell becomes "exhausted" (dysfunctional)
→ Tumor cell escapes killing
Key difference from CTLA-4:
- CTLA-4 acts early - in lymph nodes, at the costimulation stage
- PD-1/PD-L1 acts late - at the tumor site itself, on already-activated T cells
- This is why PD-1 blockade is often more targeted/less autoimmune side effects than CTLA-4 blockade
The drugs:
| Target | Drug Names | Mnemonic |
|---|---|---|
| Anti-PD-1 (blocks PD-1 on T cell) | Cemiplimab, Nivolumab, Pembrolizumab | "Nivol... Pembro... Cemip" - block the receptor |
| Anti-PD-L1 (blocks ligand on tumor/immune cell) | Atezolizumab, Durvalumab, Avelumab | "Atezo, Durva, Ave" - block the ligand |
| Anti-CTLA-4 | Ipilimumab | "Ipi" = "I block CTLA-4" |
Memory trick for PD-1 drugs: "Can Nivolumab Prevent death?" → Cemiplimab, Nivolumab, Pembrolizumab
Why Two Different Checkpoint Targets?
They work at different stages and have additive effects. Combining nivolumab (anti-PD-1) + ipilimumab (anti-CTLA-4) is now standard in melanoma and lung cancer - the combination is more effective but also more toxic. - Roitt's Essential Immunology, p. 508
Part 2: Cancer Epidemiology
Important note: Skin cancer (basal cell > squamous cell >> melanoma) is the MOST COMMON cancer overall, but it is excluded from most ranking tables because it is almost never fatal and not tracked the same way.
Cancer Incidence (who GETS it)
| Rank | Males | Females | Children (0-14) |
|---|---|---|---|
| 1 | Prostate | Breast | Leukemia |
| 2 | Lung | Lung | CNS tumors |
| 3 | Colon/Rectum | Colon/Rectum | Neuroblastoma |
Note: Lung cancer incidence has decreased in males (due to declining smoking rates) but has not significantly changed in females.
Cancer Mortality (who DIES from it)
| Rank | Males | Females | Children (0-14) |
|---|---|---|---|
| 1 | Lung | Lung | Leukemia |
| 2 | Prostate | Breast | CNS tumors |
| 3 | Colon/Rectum | Colon/Rectum | Neuroblastoma |
Key observation: Lung cancer kills the most in both sexes, even though it is not the most common cancer in males (prostate is) or females (breast is). This is because lung cancer has a very poor prognosis (often caught late).
Overall: Cancer is the 2nd leading cause of death in the United States. Heart disease is 1st.
High-Yield USMLE Memory Points
| Fact | Detail |
|---|---|
| Most common cancer overall | Skin (BCC > SCC >> melanoma) |
| Most common cancer in males | Prostate |
| Most common cancer in females | Breast |
| #1 cancer killer - both sexes | Lung |
| Most common childhood cancer | Leukemia (ALL > AML) |
| Cancer = cause of death #? | 2nd (after heart disease) |
Putting It Together: The Big Picture
The checkpoint inhibitor drugs on this page are directly linked to the immune evasion hallmark from the previous page. Tumors survive by expressing PD-L1 and exploiting CTLA-4 - checkpoint inhibitors reverse this and are now among the most important cancer treatments discovered in the past two decades, having won the 2018 Nobel Prize in Physiology or Medicine (Tasuku Honjo for PD-1, James Allison for CTLA-4).

20260715_174944.jpg
Explain
analyse_media
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
I now have everything needed. Here is a thorough explanation of page 219.
Common Metastases - Full Explanation
The Key Rule: CLaSH
The page opens with one of the most high-yield mnemonics in pathology:
Carcinomas spread via Lymphatics; Sarcomas spread Hematogenously = CLaSH
| Tumor Type | Preferred Route | Reason |
|---|---|---|
| Carcinomas (epithelial origin - lung, breast, colon, prostate, etc.) | Lymphatics first | Epithelial cells are in contact with lymphatic channels; early invasion enters lymph nodes |
| Sarcomas (mesenchymal origin - bone, muscle, fat) | Blood vessels first | Mesenchymal tissue is richly vascular but poorly lymphatic |
The 4 Exceptions - "CLaSH Breakers"
Four carcinomas behave like sarcomas and spread hematogenously rather than via lymphatics. Memorize these:
- Follicular thyroid carcinoma - invades through the thyroid capsule into blood vessels; confirmed by vascular invasion on histology. - Cummings Otolaryngology, p. 3963
- Choriocarcinoma - highly vascular trophoblastic tumor; enters blood rapidly
- Renal cell carcinoma (RCC) - notorious for growing directly into the renal vein and inferior vena cava as a tumor thrombus
- Hepatocellular carcinoma (HCC) - develops in a vascular organ with sinusoidal blood flow
Memory trick: "Foolish Children Recklessly Hitch-hike" = Follicular thyroid, Choriocarcinoma, RCC, HCC
General Rules About Metastases
- Metastasis to bone, liver, lung, and brain is MORE COMMON than primary malignancy in those organs
- Metastases typically appear as multiple lesions (vs. primary tumors which appear as a solitary lesion)
- This distinction is critical on imaging - multiple lesions in the liver or brain = think metastases first
The 4 Major Sites of Metastasis
1. Bone Metastasis
Primary tumors: Prostate, Breast >> Lung > Kidney, Colon
Memory mnemonic - "Pbig LKidney Cancer" = Prostate, Breast, Lung, Kidney, Colon
Or classically: "Lead (Pb) Kills Cancer" (Pb = Prostate + Breast = "Lead" since Pb is chemical symbol)
Location: Predilection for the axial skeleton (spine, pelvis, ribs, skull, proximal femur/humerus) - because these bones have red marrow with abundant blood supply and slow sinusoidal flow, giving circulating tumor cells more time to adhere.
Types of bone metastases - this is HIGH YIELD:
| Type | Mechanism | Example Tumors | X-ray Appearance |
|---|---|---|---|
| Blastic (osteoblastic) | Tumor stimulates osteoblasts → new bone formation | Prostate, small cell lung cancer | Dense/sclerotic (white on X-ray) |
| Lytic (osteoclastic) | Tumor activates osteoclasts → bone destruction | Kidney, colon, non-small cell lung cancer, myeloma | Punched-out holes (dark on X-ray), "moth-eaten" |
| Mixed | Both processes | Breast | Mixed dense and lucent areas |
Clinical consequences of bone metastases:
- Pain (most common symptom)
- Pathologic fractures (bone weakened by lysis)
- Hypercalcemia (from lytic destruction releasing calcium)
- Spinal cord compression (from vertebral metastases - axial skeleton predilection)
- Anemia (marrow replaced → less red cell production)
The MRI image in the textbook shows spinal metastases at T8 and L1 - classic axial skeleton involvement.
2. Liver Metastasis
Primary tumors: Colon > Breast >> Pancreas, Lung, Prostate
Why colon cancer goes to liver first: The colon drains via the portal vein directly into the liver. So colon cancer cells enter portal circulation → first capillary bed they hit = the liver ("seed and soil" + anatomical drainage). This is why colon cancer is the #1 cause of liver metastases.
Appearance: Scattered throughout liver parenchyma - multiple white/pale nodules on gross pathology (as seen in the liver photo on the page - the bright white nodules against the dark liver tissue).
Clinical clues:
- Elevated ALP, GGT (cholestatic pattern)
- Hepatomegaly, right upper quadrant pain
- "Cannon ball" nodules on imaging in aggressive cases (eg, RCC, choriocarcinoma)
3. Lung Metastasis
Primary tumors: Colon, Breast >> Kidney, Prostate
Key feature: Typically involve BOTH lungs - bilateral pulmonary nodules on imaging = metastatic disease until proven otherwise.
Why? Tumor cells enter systemic venous circulation → right heart → pulmonary capillaries (first capillary bed) → lodge in both lungs.
Appearance on CXR/CT: "Cannonball" metastases - multiple rounded nodules of varying sizes scattered bilaterally. Classic appearance for RCC lung mets.
4. Brain Metastasis
Primary tumors: Lung > Breast >> Melanoma > Colon, Prostate
Key feature: Usually seen at the gray/white matter junction (corticomedullary junction)
Why the gray-white junction?
- Blood vessels narrow abruptly at the interface between cortex (gray matter) and white matter
- Tumor emboli slow down and lodge here, like debris caught at a bottleneck
- This is why brain mets appear as multiple discrete nodules right at this junction
Lung cancer is the #1 cause of brain metastases - even in patients who present with a brain lesion first, lung should be the primary suspected source (always get a chest CT).
Melanoma is notable because despite being less common than breast or colon cancer, it has an extremely high rate of brain tropism - melanoma cells have special affinity for brain tissue.
Clinical: Headache (worse in morning - due to elevated ICP), seizures, focal neurologic deficits, personality change.
Summary Table - All Metastatic Sites
| Site | Top Primary Tumors | Key Feature |
|---|---|---|
| Bone | Prostate, Breast >> Lung > Kidney, Colon | Axial skeleton; Blastic (prostate) vs Lytic (kidney) vs Mixed (breast) |
| Liver | Colon > Breast >> Pancreas, Lung, Prostate | Portal drainage explains colon dominance; multiple scattered nodules |
| Lung | Colon, Breast >> Kidney, Prostate | Both lungs involved; "cannonball" nodules |
| Brain | Lung > Breast >> Melanoma > Colon, Prostate | Gray-white matter junction; Lung = #1 cause |
High-Yield USMLE Integration
| Clinical Scenario | Think |
|---|---|
| 65M with back pain, elevated PSA, sclerotic vertebral lesions | Prostate → blastic bone mets |
| 55F with breast cancer history, new lytic hip lesion | Breast → lytic bone mets |
| Multiple bilateral lung nodules | Metastatic disease (colon, breast, RCC) |
| Multiple liver nodules in a patient with colon cancer | Liver mets via portal drainage |
| Multiple brain lesions at corticomedullary junction | Mets - check for lung primary first |
| Renal vein tumor thrombus on CT | RCC - hematogenous spreader |
| Follicular thyroid cancer with distant mets | Hematogenous - bone, lung |
| Single lung nodule vs multiple | Single = possibly primary; Multiple = metastases |
This is a shared conversation. Sign in to Orris to start your own chat.