Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathophysiology
Reading File
Reading File
Searching Images
ARDS acute respiratory distress syndrome pathophysiology mechanism diagram
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Searching the Web
ARDS mechanism pathophysiology 2024 neutrophil alveolar-capillary barrier cytokine storm
Loading Image
I now have comprehensive information from multiple authoritative sources. Let me compile the full explanation.
Acute Respiratory Distress Syndrome (ARDS): Mechanism
Definition & Diagnostic Criteria (Berlin Definition)
ARDS is a syndrome of non-cardiogenic pulmonary edema driven by diffuse inflammatory lung injury. The Berlin criteria require:
- Bilateral infiltrates on CXR or CT (not fully explained by pleural effusions, atelectasis, or nodules)
- PaO₂/FiO₂ ratio < 300 (mild: 200–300; moderate: 100–200; severe: <100) with PEEP ≥ 5 cm H₂O
- Onset within 7 days of a known clinical insult
- Not fully explained by heart failure or fluid overload
Common triggers include sepsis, pneumonia, aspiration, trauma, massive transfusion, pancreatitis, and burns.
Core Pathophysiology: The Alveolar-Capillary Barrier
The central event in ARDS is breakdown of the alveolar-capillary barrier — the normally tight interface between alveolar epithelium (type I and II pneumocytes) and pulmonary capillary endothelium. Under physiological conditions, tight junctions and surfactant maintain barrier integrity. In ARDS, this barrier is disrupted by:
- Direct injury (e.g., inhaled toxins, gastric aspiration, pneumonia)
- Indirect/systemic injury (e.g., sepsis-mediated mediator release reaching the lung via the bloodstream)
Step-by-Step Mechanistic Cascade
1. Initiating Insult → Innate Immune Activation
An insult — infection, trauma, or ischemia — activates pattern recognition receptors (PRRs) including Toll-like receptors (TLRs) on alveolar macrophages. Macrophages polarize to the M1 phenotype, releasing a cascade of pro-inflammatory cytokines:
- TNF-α, IL-1β, IL-6, IL-8 (CXCL8)
These cytokines activate the NF-κB and STAT3 signaling pathways, amplifying the inflammatory signal.
2. Cytokine Storm
The rapid, excessive release of cytokines and chemokines constitutes a cytokine storm — a hallmark of ARDS. Key effects:
| Cytokine | Effect |
|---|---|
| TNF-α, IL-1β | Endothelial activation, increased vascular permeability |
| IL-6 | Systemic inflammation, acute-phase response |
| IL-8 (CXCL8) | Potent neutrophil chemoattractant |
| IL-17 (via Th17 cells) | Amplifies neutrophil recruitment |
Simultaneously, anti-inflammatory mediators (IL-10, TGF-β via T-regulatory cells) are suppressed, removing the normal brake on inflammation.
3. Neutrophil Recruitment & Activation — The Central Effector
Neutrophils are the principal effector cells of lung injury. IL-8 and other chemokines (CCL1, CXCL1) drive massive neutrophil sequestration in the alveolar and interstitial spaces. Activated neutrophils release:
- Reactive oxygen species (ROS) and reactive nitrogen species (RNS) → oxidative damage to epithelium and endothelium
- Proteases (elastase, matrix metalloproteinases) → degrade basement membranes and tight junctions
- Myeloperoxidase (MPO) → further oxidative injury
- Neutrophil extracellular traps (NETs) → ensnare pathogens but also damage endothelial cells and trigger immunothrombosis
Platelet–neutrophil complexes form via P-selectin/PSGL-1 interactions, further amplifying ROS and NET release and promoting microvascular thrombosis.
4. Alveolar Epithelial Injury
- Type I pneumocytes (covering ~95% of alveolar surface) are highly susceptible — their destruction floods the alveolus with protein-rich edema fluid
- Type II pneumocyte injury has compounding effects:
- Loss of surfactant production → alveolar collapse and dramatically increased surface tension
- Loss of alveolar repair capacity (type II cells are the progenitors of type I)
- Impaired active ion transport (Na⁺-K⁺-ATPase) → failure of edema clearance
5. Pulmonary Endothelial Injury → Non-Cardiogenic Edema
TNF-α, IL-1β, and direct cellular toxins disrupt VE-cadherin and other endothelial junctional proteins, enlarging inter-endothelial gaps. The result:
- Protein-rich exudate floods the alveolar space (pulmonary edema with high protein content — unlike the transudate of cardiogenic edema)
- Hydrostatic pressure is normal (wedge pressure < 18 mmHg), distinguishing ARDS from heart failure
- Fibrinogen enters the alveolus and polymerizes into hyaline membranes — a pathological hallmark on histology (diffuse alveolar damage, DAD)
6. Surfactant Failure → Atelectasis
Phospholipase A₂ (elevated in pancreatitis-associated ARDS) enzymatically degrades surfactant phospholipids. Combined with type II pneumocyte loss, surfactant is both degraded and under-produced. This causes:
- Progressive alveolar collapse (atelectasis)
- V/Q mismatch and intrapulmonary shunting
- Refractory hypoxemia (the defining clinical feature)
7. Coagulation Dysregulation
- Pro-inflammatory cytokines trigger tissue factor expression on endothelium → local fibrin deposition and microthrombi
- Simultaneously, fibrinolysis is impaired (elevated PAI-1)
- Result: immunothrombosis in pulmonary microvasculature → further V/Q mismatch and dead-space physiology
Phases of ARDS
| Phase | Timing | Pathology |
|---|---|---|
| Exudative | Days 1–7 | Neutrophil influx, alveolar flooding, hyaline membrane formation, DAD |
| Proliferative | Days 7–21 | Type II pneumocyte proliferation, early fibroblast infiltration, organization of exudate |
| Fibrotic | Weeks–months | Collagen deposition, architectural distortion, obliteration of alveolar units (in ~30% of patients) |
Mechanical Ventilation as a Secondary Injury (Ventilator-Induced Lung Injury, VILI)
Because ARDS creates heterogeneous lung injury (some alveoli flooded, some atelectatic, some relatively normal), conventional tidal volumes cause volutrauma and barotrauma in the small fraction of "baby lung" units that remain open. This worsens injury via:
- Cyclic stretch → mechanical activation of NF-κB → further cytokine release
- Atelectrauma (repetitive opening and closing of unstable alveoli)
- Biotrauma → cytokines leak from the lung into the systemic circulation → MODS (multiple organ dysfunction syndrome)
This is why lung-protective ventilation (6–8 mL/kg predicted body weight, plateau pressure ≤30 cm H₂O) is the only intervention with proven mortality benefit.
Gravitational Distribution of Injury
The edematous ARDS lung behaves like a sponge — edema fluid preferentially fills dependent zones (posterior in supine patients), producing:
- Dependent zones: consolidated/fluid-filled, contributing to shunt
- Non-dependent zones: relatively aerated but small ("baby lung")
- Intermediate zones: atelectatic but potentially recruitable
This explains why prone positioning improves oxygenation and reduces mortality — it redistributes perfusion to better-ventilated regions.
Pathophysiology Diagram
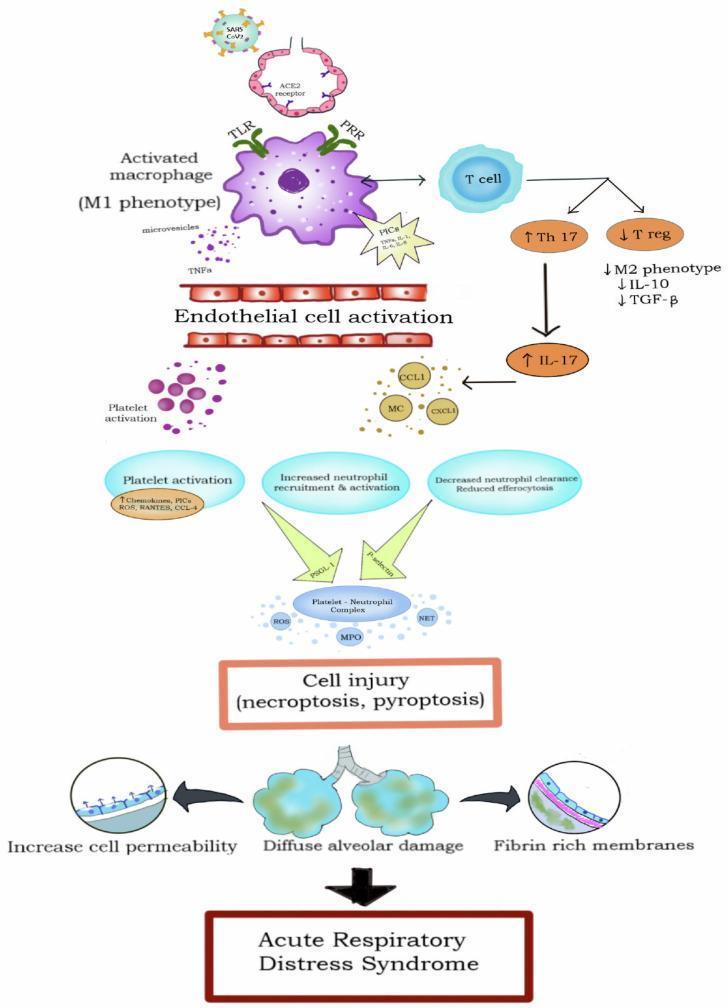
Summary: Key Mechanistic Steps
Initiating insult (sepsis, pneumonia, trauma)
↓
Macrophage activation (TLR/PRR) → Cytokine storm (TNF-α, IL-1β, IL-6, IL-8)
↓
Neutrophil recruitment → ROS, proteases, NETs
↓
Alveolar-capillary barrier disruption
├── Endothelial injury → Protein-rich edema floods alveoli
├── Type I pneumocyte loss → Loss of gas exchange surface
└── Type II pneumocyte loss → Surfactant failure + impaired repair
↓
Hyaline membrane formation (diffuse alveolar damage)
↓
Surfactant deficiency → Alveolar collapse → V/Q mismatch → Shunt
↓
Refractory hypoxemia + reduced lung compliance (↓ΔV/ΔP)
↓
If uncontrolled → Fibroproliferative phase → Fibrosis / MODS
Sources: Murray & Nadel's Textbook of Respiratory Medicine; Goldman-Cecil Medicine; Sabiston Textbook of Surgery; Nature Signal Transduction & Targeted Therapy 2025; Frontiers Cell & Developmental Biology 2025
This is a shared conversation. Sign in to Orris to start your own chat.