Cystic fibrosis Detailed note for clinical cardio 15 marks bpt mgr university exam
Reading File
Finding Sources
Finding Sources
Searching the Web
cystic fibrosis clinical features pathophysiology cardiovascular complications BPT exam notes
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Reading File
Excellent! I now have comprehensive information from multiple authoritative textbooks. Let me compile the complete 15-mark detailed note for a BPT MGR University Clinical Cardiology exam.
CYSTIC FIBROSIS - Detailed Clinical Note (15 Marks)
BPT MGR University | Clinical Cardiology
1. DEFINITION
Cystic fibrosis (CF) is an autosomal recessive multisystem disorder caused by mutations in the CFTR (Cystic Fibrosis Transmembrane Conductance Regulator) gene, which encodes a chloride and bicarbonate channel on the apical surface of epithelial cells. The dysfunction of this channel leads to abnormally thick, viscid secretions affecting the airways, pancreas, liver, intestines, reproductive tract, and sweat glands.
- Robbins & Kumar Basic Pathology - the CFTR protein resides on the surface of epithelial cells in the airways, pancreatic ducts, biliary tree, intestine, vas deferens, and sweat glands, resulting in progressive obstructive pulmonary disease.
2. EPIDEMIOLOGY
| Parameter | Detail |
|---|---|
| Incidence | 1 in 2000-2500 live births in Caucasians |
| Incidence (African Americans) | 1 in 17,000 |
| Inheritance | Autosomal recessive |
| Gene locus | Chromosome 7q31.2 |
| Commonest mutation | ΔF508 (deletion of phenylalanine at position 508) - ~70% in Northern Europeans |
| Median survival | Now extended beyond 47 years (with modern therapies) |
3. MOLECULAR PATHOGENESIS AND CFTR MUTATIONS
The CFTR gene was cloned in 1989. Over 2000 disease-associated mutations have been identified. Mutations are classified into 6 classes based on their effect on CFTR protein (Robbins, Cotran & Kumar):
| Class | Defect | Example |
|---|---|---|
| I | Defective protein synthesis (null mutations) - premature stop codon, no CFTR at apical surface | G542X, W1282X |
| II | Abnormal protein folding, processing, and trafficking - protein degraded before reaching cell surface | ΔF508 (most common) |
| III | Defective regulation (gating mutations) - normal CFTR at surface but nonfunctional; ATP binding impaired | G551D |
| IV | Decreased conductance - CFTR present but reduced Cl⁻ conduction through transmembrane domain | R117H |
| V | Reduced abundance - splice-site or promoter mutations; reduced amount of normal protein | - |
| VI | Decreased membrane stability - functional protein but greatly reduced stability at membrane | - |
- Classes I-III = "severe" mutations (<10% residual CFTR activity)
- Classes IV-VI = "mild" mutations (<20% residual CFTR activity)
Genotype-Phenotype Correlation
Two severe mutations → classic CF phenotype (pancreatic insufficiency, sinopulmonary infections, GI symptoms). One mild + one severe → less severe phenotype. Genotype-phenotype correlation is most consistent for pancreatic disease; pulmonary severity is influenced by additional environmental modifiers (e.g., Pseudomonas colonization).
4. PATHOPHYSIOLOGY
The CFTR channel normally regulates chloride secretion and sodium absorption across epithelial cells. In CF:
- Loss of CFTR function → failure of Cl⁻ and HCO₃⁻ secretion → reduced surface liquid hydration
- Compensatory hyperabsorption of Na⁺ (via ENaC channels) → water follows → dehydration of periciliary fluid
- Airway surface liquid depletion → thick, viscid mucus → impaired mucociliary clearance
- Mucus stasis → bacterial colonization (S. aureus early; Pseudomonas aeruginosa chronically - mucoid strains)
- Chronic infection → neutrophilic inflammation → proteases (neutrophil elastase) → airway wall destruction → bronchiectasis
- Sweat glands: CFTR dysfunction → failure to reabsorb Cl⁻ → elevated sweat chloride (diagnostic hallmark)
- Pancreatic ducts: viscid secretions obstruct ducts → acinar atrophy, fibrosis → exocrine pancreatic insufficiency (85-90% of patients)
5. CLINICAL FEATURES
The clinical manifestations are multisystemic. The diagram below from Harriet Lane Handbook illustrates the breadth:
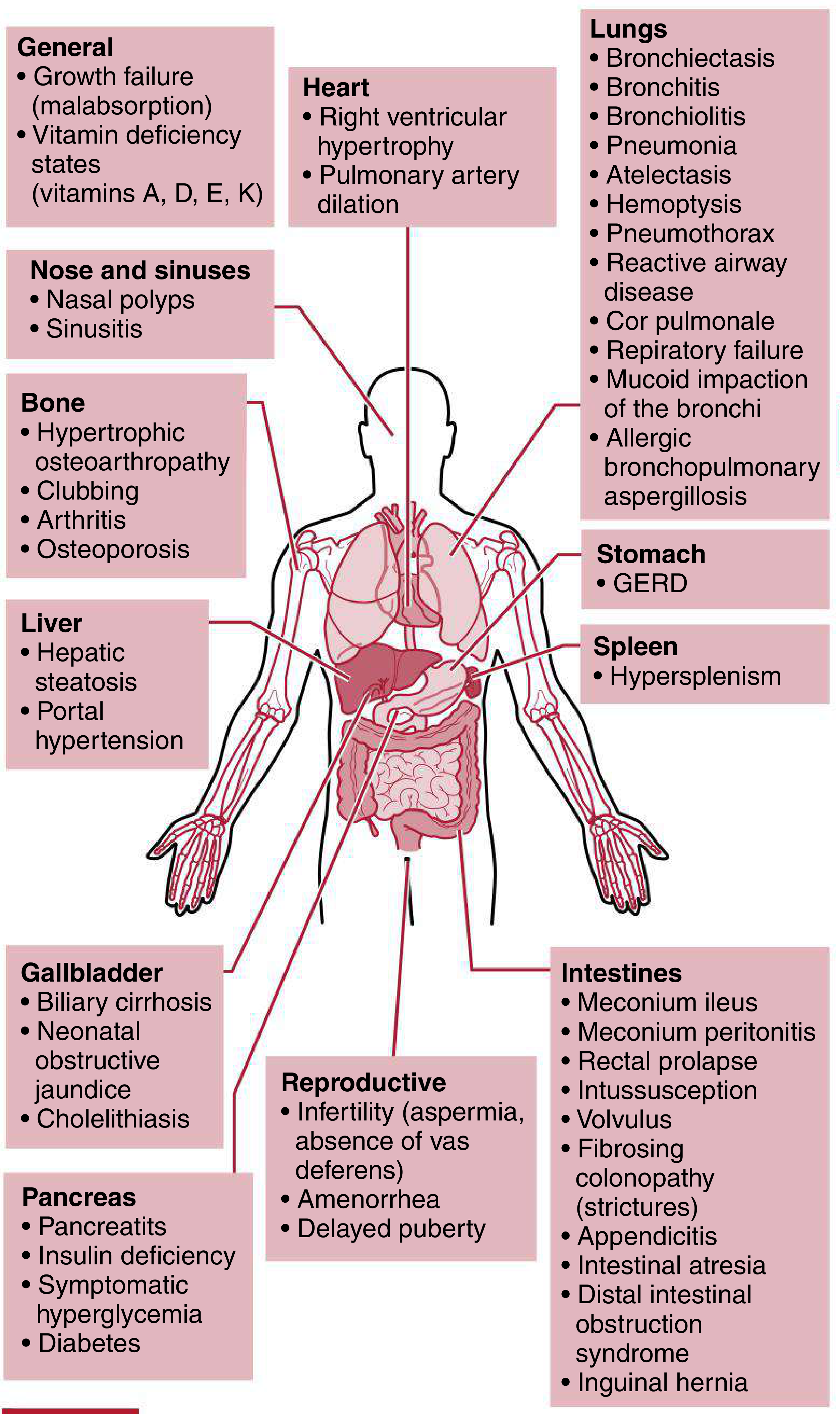
A. PULMONARY (90% of patients)
- Chronic productive cough with thick, purulent sputum
- Recurrent respiratory tract infections (S. aureus, H. influenzae, mucoid P. aeruginosa)
- Progressive obstructive lung disease
- Bronchiectasis - the hallmark structural complication
- Bronchitis, bronchiolitis, atelectasis
- Hemoptysis (small streaks common; massive hemoptysis in advanced disease)
- Spontaneous pneumothorax
- Reactive airway disease / wheeze
- Mucoid impaction of bronchi
- Allergic bronchopulmonary aspergillosis (ABPA)
- Respiratory failure - most common cause of death
B. CARDIOVASCULAR (Clinical Cardiology Focus)
- Cor Pulmonale - right heart failure secondary to pulmonary hypertension; results from:
- Chronic hypoxemia → hypoxic pulmonary vasoconstriction
- Destruction of pulmonary capillary bed from bronchiectasis and fibrosis
- Leads to right ventricular hypertrophy and dilation
- Pulmonary artery dilation
- Pulmonary hypertension - a late complication in advanced CF
- Sildenafil has been used for CF-related pulmonary hypertension (limited evidence)
- Cardiac arrhythmias and right heart failure as terminal events
C. UPPER RESPIRATORY TRACT
- Chronic sinusitis (nearly universal)
- Nasal polyps (10-50%)
- Rhinosinusitis
D. GASTROINTESTINAL
- Meconium ileus in neonates (10-25%) - 95% of meconium ileus cases have underlying CF
- Distal intestinal obstruction syndrome (DIOS) - adult equivalent of meconium ileus (18% of adults)
- Rectal prolapse (1-2%)
- Intussusception
- Volvulus
- Fibrosing colonopathy (intestinal strictures)
- GERD (80% of adults)
E. PANCREATIC
- Exocrine pancreatic insufficiency (85-90%) → malabsorption, steatorrhea
- Pancreatitis (1-2%)
- Cystic fibrosis-related diabetes mellitus (4-7%) - unique mixed type (insulin deficiency + resistance)
- Abnormal glucose tolerance (20-30%)
F. HEPATOBILIARY
- Hepatic steatosis (fatty liver) - 7-20%
- Focal biliary cirrhosis (in ~one-third of patients over time)
- Portal hypertension (2-3%, up to 28% in adults)
- Neonatal obstructive jaundice
- Gallstones (8-25%)
- Hypersplenism
G. MUSCULOSKELETAL
- Hypertrophic pulmonary osteoarthropathy (HPOA) - clubbing of fingers and toes
- Arthritis
- Osteoporosis / osteopenia (due to malabsorption of fat-soluble vitamins D and K, and chronic inflammation)
H. REPRODUCTIVE
- Males: Infertility due to congenital bilateral absence of vas deferens (CBAVD) - azoospermia (~98% of CF males)
- Females: Reduced fertility due to thick cervical mucus, delayed puberty from nutritional deficits; ~4% of female CF patients aged 17-37 may be pregnant at any time
- Amenorrhea, delayed puberty
I. SWEAT GLANDS
- Salty sweat - parents may notice salty taste when kissing child
- Excessive salt loss → dehydration, hyponatremia, metabolic alkalosis in infants
J. GENERAL
- Growth failure / failure to thrive (malabsorption)
- Vitamin deficiency (A, D, E, K - fat-soluble vitamins)
6. MORPHOLOGY / PATHOLOGICAL CHANGES
Based on Robbins, Cotran & Kumar:
| Organ | Pathological Changes |
|---|---|
| Lungs | Mucus plugging of airways, submucosal gland hypertrophy, bronchiectasis, bronchial wall thickening, lung hyperinflation |
| Pancreas | Mucus accumulation in small ducts → ductal dilation → acinar atrophy → progressive fibrosis → only islets remain in fibrofatty stroma |
| Liver | Bile canaliculi plugged by mucus → ductular proliferation → portal inflammation → focal biliary cirrhosis (1/3 of patients) |
| Salivary glands | Ductal dilation, squamous metaplasia, glandular atrophy, fibrosis |
| Intestine | Thick viscid mucus plugs → meconium ileus in newborns |
| Sweat glands | Morphologically unaffected (functional defect only) |
| Heart | Right ventricular hypertrophy (cor pulmonale), pulmonary artery dilation |
7. DIAGNOSIS
A. Newborn Screening (NBS)
- Blood immunoreactive trypsinogen (IRT) level - elevated in CF due to blocked pancreatic ducts
- CFTR gene mutation analysis on dried blood spot
B. Sweat Chloride Test (Gold Standard)
- Quantitative pilocarpine iontophoresis - stimulates sweating
- Diagnostic: Cl⁻ >60 mmol/L (two separate occasions)
- Borderline: 30-59 mmol/L
- Normal: <30 mmol/L
- False positives: adrenal insufficiency, hypothyroidism, nephrogenic DI, ectodermal dysplasia, malnutrition, glycogen storage disease type I
C. Genetic Analysis
- Over 2500 CFTR mutations described
- CFTR gene sequencing if mutation not identified on screening panel
- Two disease-causing CFTR variants in trans configuration = diagnostic
D. Pulmonary Function Tests (PFTs)
- Obstructive pattern (reduced FEV1, FEV1/FVC ratio)
- FEV1 used to track disease severity and predict outcomes
- Decline in FEV1 = most reliable marker of disease progression
E. Chest X-Ray / CT Chest
- Hyperinflation, peribronchial thickening, mucus plugging
- Bronchiectasis (ring shadows, tram-tracks)
- Upper lobe predominance in CF bronchiectasis
F. Additional Tests
- Sputum culture and sensitivity (Pseudomonas aeruginosa, MRSA)
- Nasal potential difference measurement (CFTR function)
- Pancreatic function: fecal elastase, 72-hour fecal fat
8. MANAGEMENT
A. Pulmonary - Airway Clearance
- Chest physiotherapy (CPT) - percussion and postural drainage (hallmark BPT intervention)
- High-frequency chest wall oscillation (HFCWO) - "Vest" device
- Active cycle of breathing technique (ACBT)
- Oscillatory PEP devices (Flutter, Acapella)
B. Aerosolized Agents
- Dornase alfa (DNase) - recombinant human DNase; cleaves extracellular DNA from neutrophils, reducing mucus viscosity
- Hypertonic saline (7%) - hydrates airway mucus, stimulates cough, improves mucociliary clearance
C. Antibiotics
- Acute exacerbations: IV antibiotics targeting Pseudomonas (anti-pseudomonal penicillins, aminoglycosides, fluoroquinolones)
- Chronic Pseudomonas colonization: aerosolized tobramycin or aztreonam alternating monthly
- Chronic azithromycin - reduces exacerbation frequency (immunomodulatory effect)
D. CFTR Modulator Therapy (Targeted Therapy)
| Drug | Mechanism | Mutation |
|---|---|---|
| Ivacaftor (Kalydeco) | Potentiator - opens CFTR gate | G551D (Class III) |
| Lumacaftor + Ivacaftor | Corrector + potentiator | ΔF508 homozygous |
| Tezacaftor + Ivacaftor | Corrector + potentiator | ΔF508 |
| Elexacaftor + Tezacaftor + Ivacaftor (Trikafta) | Triple therapy (highly effective) | ΔF508 (one or two alleles) |
Ivacaftor trial showed ~50% reduction in sweat chloride, significant FEV1 improvement, fewer exacerbations, and weight gain.
E. Nutritional Management
- High-calorie, high-fat diet (120-150% of normal caloric requirements)
- Pancreatic enzyme replacement therapy (PERT) - lipase + amylase + protease capsules with meals
- Fat-soluble vitamin supplementation: A, D, E, K
- Management of CF-related diabetes: insulin therapy
F. Management of Cardiovascular Complications
- Cor pulmonale: supplemental O₂ therapy (mainstay), diuretics (cautiously), pulmonary vasodilators (sildenafil - limited evidence)
- Optimize pulmonary treatment to reduce hypoxemia
- Avoid beta-blockers (may worsen bronchospasm)
- Lung transplantation - definitive therapy in end-stage disease; bilateral sequential lung transplantation improves survival
G. Lung Transplantation
- Indicated for end-stage CF (FEV1 <30%, rapid decline, recurrent life-threatening hemoptysis, or hypercapnia)
- Bilateral sequential lung transplant is the procedure of choice
- 5-year survival post-transplant ~50%
9. CARDIOVASCULAR MANIFESTATIONS IN DETAIL (Cardiology Focus)
Since this is a Clinical Cardiology paper, this section deserves special emphasis:
Cor Pulmonale in CF
- CF is listed as a predisposing disease to cor pulmonale (Robbins & Kumar - Table 9.3 - Disorders Predisposing to Cor Pulmonale includes: COPD, Diffuse pulmonary interstitial fibrosis, Pneumoconiosis, Cystic fibrosis, Bronchiectasis)
Mechanism:
- Chronic hypoxemia from obstructive lung disease and V/Q mismatch
- Hypoxic pulmonary vasoconstriction → elevated pulmonary vascular resistance
- Destruction of pulmonary capillary bed by bronchiectasis, fibrosis, and emphysema
- Pulmonary artery remodeling → fixed pulmonary hypertension
- Right ventricle works against chronically elevated afterload → RV hypertrophy → eventually RV failure
Clinical signs of cor pulmonale in CF:
- Elevated JVP
- Right ventricular heave
- Loud P2 (pulmonary component of S2)
- Tricuspid regurgitation murmur
- Peripheral edema
- Hepatomegaly (congestive)
Echocardiographic findings:
- RV hypertrophy and dilation
- Tricuspid annular plane systolic excursion (TAPSE) reduced
- Elevated RVSP (right ventricular systolic pressure)
- Pulmonary artery dilation
Management of cor pulmonale in CF:
- Supplemental O₂ (target SpO₂ >90%) - reduces hypoxic vasoconstriction
- Optimal CF pulmonary treatment (antibiotics, airway clearance, modulators)
- Diuretics: furosemide - reduce preload, edema
- Sildenafil (PDE-5 inhibitor): used for CF-related pulmonary hypertension (Goldman-Cecil Medicine)
- Lung transplantation - only definitive therapy
10. PROGNOSIS
- Median survival has dramatically improved: now >47 years in many centers (Sleisenger & Fordtran)
- Most common cause of death: respiratory failure (>90% of deaths)
- Poor prognostic indicators: FEV1 <30%, chronic Pseudomonas colonization, malnutrition, frequent exacerbations, diabetes, female sex (historically)
- CFTR modulator therapy (especially Trikafta) has transformed outcomes - significant improvement in FEV1, nutritional status, quality of life, and reduction in hospitalizations
11. SUMMARY TABLE
| Feature | Detail |
|---|---|
| Gene | CFTR, chromosome 7q31.2 |
| Commonest mutation | ΔF508 (Class II) |
| Inheritance | Autosomal recessive |
| Diagnostic test | Sweat Cl⁻ >60 mmol/L (gold standard) |
| Lungs | Bronchiectasis, chronic Pseudomonas infection, obstructive pattern |
| Cardiovascular | Cor pulmonale, RV hypertrophy, pulmonary hypertension |
| Pancreas | Exocrine insufficiency (85-90%), CF-related diabetes |
| GI | Meconium ileus (neonate), DIOS (adult) |
| Reproductive | Azoospermia (males), reduced fertility (females) |
| Key treatment (BPT) | Chest physiotherapy + postural drainage |
| Targeted therapy | CFTR modulators (Ivacaftor, Trikafta) |
| Cause of death | Respiratory failure |
Sources:
- Robbins, Cotran & Kumar: Pathologic Basis of Disease - Chapter 10 (Cystic Fibrosis genetics and morphology)
- Harriet Lane Handbook, 23rd ed. - Chapter 25 (Clinical manifestations and management)
- Murray & Nadel's Textbook of Respiratory Medicine - Chapters 16, 30
- Sleisenger & Fordtran's GI and Liver Disease - Table 57.3 (GI manifestations frequency)
- Robbins & Kumar Basic Pathology - Table 9.3 (Cor pulmonale predisposing disorders)
- Goldman-Cecil Medicine - Pulmonary hypertension management in CF
This is a shared conversation. Sign in to Orris to start your own chat.