Management of c section pt
Management of the Cesarean Section Patient
1. Indications
- Dystocia: fetopelvic disproportion, abnormal fetal presentation (breech, transverse/oblique lie), dysfunctional uterine activity
- Fetal distress: non-reassuring fetal heart rate, umbilical cord prolapse with fetal bradycardia
- Maternal hemorrhage: placenta previa (central/partial), abruptio placentae
- Previous uterine surgery: prior classic cesarean section, extensive myomectomy
- Maternal conditions: active genital herpes with ruptured membranes, impending maternal death
- Repeat cesarean delivery
2. Pre-Operative Management
Airway and Aspiration Prevention
- All pregnant patients after 20 weeks are at increased risk of pulmonary aspiration due to reduced lower esophageal sphincter competence and potential delayed gastric emptying in labor
- Administer a non-particulate antacid (e.g., sodium citrate) within 30 minutes of anticipated surgery
- Consider H2 blockers or metoclopramide in high-risk patients
- Regional anesthesia is preferred over general anesthesia to reduce aspiration risk
IV Access and Fluid Preloading
- Establish large-bore IV access
- Administer IV fluid co-load (crystalloid bolus at the time of induction of neuraxial anesthesia is as effective as pre-loading)
- Cross-match blood for emergencies
Patient Positioning
- Left lateral uterine displacement to relieve aortocaval compression and prevent supine hypotension
Antibiotic Prophylaxis
- Prophylactic antibiotics before skin incision reduce wound infection to <3% of cesarean deliveries
- Adding azithromycin (a macrolide) to standard cephalosporin prophylaxis for non-elective cesarean delivery significantly reduces endometritis and wound infection rates (Creasy & Resnik's Maternal-Fetal Medicine)
3. Anesthesia Management
Regional Anesthesia (Preferred)
- Requires sensory block up to T4 dermatome
- Advantages: rapid, predictable onset; denser block; less systemic drug toxicity; no fetal drug depression; allows spinal opioids for post-op pain; mother is awake for the birth
- Spinal drug: hyperbaric bupivacaine is standard; intrathecal opioids (morphine, fentanyl) added for post-op analgesia
- Slower onset but allows more controlled level titration
- Preferred when an epidural is already in situ from labor
- Combines the reliability of spinal with the flexibility of epidural for prolonged procedures
- Monitor BP every 2-3 minutes after induction
- Phenylephrine is preferred over ephedrine - equally efficacious but results in less fetal acidosis; given as bolus (100-150 mcg) or prophylactic infusion
- Norepinephrine (bolus 6 mcg IV) is an alternative - has beta-adrenergic effects reducing risk of reflex bradycardia
General Anesthesia (Emergency/Regional Contraindicated)
- Rapid-sequence induction (RSI) with cricoid pressure
- Denitrogenation: 3-5 minutes with 100% O2, or 4 deep breaths in emergencies
- Induction: propofol 2 mg/kg, ketamine 1 mg/kg, or etomidate 0.2-0.3 mg/kg
- Muscle relaxant: succinylcholine 1-1.5 mg/kg or rocuronium 1.0 mg/kg (reversible with sugammadex)
- Maintenance: 50:50 N2O/O2 + volatile agent
- Confirm ETT position with capnography and auscultation before skin incision
- Key risk: failed intubation - follow the OAA/Difficult Airway Society algorithm
4. Intra-Operative Surgical Management
Incision Types
- Pfannenstiel (transverse) incision: most common for elective cases; better cosmesis, fewer complications
- Vertical (midline) incision: for emergencies or when rapid access is needed; allows better surgical exposure
Uterine Incision
- Low transverse (Kerr) uterine incision: standard - lower blood loss, lower risk of uterine rupture in subsequent pregnancies, excluded from peritoneal cavity reducing sepsis risk
- Classical (vertical) uterine incision: reserved for transverse lie, extreme prematurity, or anterior placenta previa
Delivery
- Delivery of infant from cephalic or breech presentation
- Vacuum extractor may be used to assist delivery
- Delivery of placenta (manual or spontaneous)
- Closure of uterus and abdomen in layers
5. Post-Operative Management
Pain Management
- Neuraxial opioids: intrathecal morphine provides 12-24 hours of post-op pain relief; preferred first-line
- NSAIDs + acetaminophen: systemic, scheduled
- TAP block (Transversus Abdominis Plane) or Quadratus Lumborum (QL) block: indicated when neuraxial morphine was not given or is insufficient; especially for patients on buprenorphine
- PCEA (patient-controlled epidural analgesia): with dilute local anesthetic + lipid-soluble opioid for patients with high pain risk
Monitoring and Routine Post-Operative Care
- Vital signs monitoring, urine output via Foley catheter
- Encourage early ambulation (reduces DVT risk)
- VTE prophylaxis: DVT occurs in 1-2% of cesarean deliveries; risk factors include obesity, prolonged surgery, endometritis, thrombophilia
- Prophylactic heparin (LMWH or UFH) + compression stockings
- Pulmonary embolism is a leading cause of maternal mortality
Wound Care
- Prophylactic antibiotics reduce wound infection to <3%
- Risk factors for wound infection: second-stage cesarean, obesity, suprafascial drains
- 25-30% of wound infections are due to Staphylococcus aureus (not endometrial flora) - sterile technique is essential
- Wound hematoma: due to faulty hemostasis, managed with drainage
6. Complications and Their Management
Endomyometritis
- Polymicrobial infection; symptoms: fever, uterine tenderness, abdominal pain, malodorous lochia
- Treatment: IV clindamycin + gentamicin (most effective combination per systematic review of 47 trials)
- Regimens active against penicillin-resistant anaerobes are superior
- Oral follow-up antibiotics not needed once clinical improvement occurs
Urinary Tract Complications
- Urinary tract injuries: 10x more common in cesarean than operative vaginal delivery; most are bladder lacerations, repaired intraoperatively
- Bacteriuria: ~11% of cases, related to urinary catheterization
- Reduce UTI risk by eliminating or using intermittent rather than indwelling catheterization
Thromboembolic Disease
- Puerperal DVT: 1-2% of cesarean deliveries
- Treatment: IV heparin followed by oral anticoagulants
- Ovarian vein thrombosis: right-sided tender palpable mass extending to the flank - a rare but recognized complication
- Pelvic thrombophlebitis: diagnosed by exclusion when fever doesn't respond to antibiotics but resolves with heparin
Respiratory Complications
- Atelectasis: most common early post-op complication; treated with incentive spirometry, early ambulation
- Neonatal respiratory morbidity is increased with elective pre-labor cesarean, especially at 37-38 weeks (13-38 fold increased risk vs 39-40 weeks) - hence elective cesareans should not be performed before 39 weeks gestation without medical indication
7. Future Pregnancy Considerations (Long-term Complications)
- Increased risk of placenta previa in subsequent pregnancies (OR 1.4)
- Increased risk of placenta accreta with multiple prior cesareans
- Possible increased infertility (may relate to fallopian tube damage from infection)
- Uterine scar - higher risk of uterine rupture in subsequent labor (especially with classical incision)
- Creasy & Resnik's Maternal-Fetal Medicine, pp. 958-961 (Cesarean Delivery and Complications)
- Morgan & Mikhail's Clinical Anesthesiology, 7e (Anesthesia for Cesarean Section)
- Barash, Cullen & Stoelting's Clinical Anesthesia, 9e (Postoperative Analgesia & Anesthetic Complications)
What about diet, postural after post-op and, um, other general management or specific management with the causology and pathophysiology in details for a caesarean section patient in a primary?
enhanced recovery after cesarean section ERAS protocol
PMID: 38042118
cesarean section postoperative management diet posture primary care evidence-based 2024
https://www.ajog.org/article/S0002-9378(25)00071-7/fulltext
https://www.ajog.org/article/S0002-9378(25)00071-7/fulltext
cesarean section pathophysiology wound healing VTE risk bowel ileus physiological recovery
Post-Operative Management of the Cesarean Section Patient (Primary Care Focus)
Understanding the Pathophysiology After Cesarean Section
Surgical Stress Response
- The hypothalamic-pituitary-adrenal axis activates, releasing cortisol and catecholamines
- This triggers a catabolic, hypermetabolic state: protein is broken down for gluconeogenesis, wound repair draws on nutritional stores, and immune function is temporarily suppressed
- Tissue trauma releases pro-inflammatory cytokines (IL-1, IL-6, TNF-alpha), causing systemic inflammation, fever, and edema in the early post-op period
Wound Healing Phases
- Haemostasis (seconds - minutes): vasoconstriction, platelet plug, fibrin clot formation
- Inflammation (0-4 days): neutrophils and macrophages clear debris; this causes expected warmth, redness, and swelling at the wound site
- Proliferation (2-24 days): fibroblasts lay down collagen, granulation tissue forms, epithelialisation occurs
- Remodelling (24 days - 1 year): collagen reorganises; tensile strength gradually restores
Uterine Physiology Post-Delivery
- The uterus undergoes involution: from ~1 kg at term back to ~60 g over 6 weeks
- Fundal height decreases approximately 1 cm/day
- Lochia progresses: rubra (red, days 1-4) → serosa (pink/brown, days 4-10) → alba (white/yellow, days 10-28)
- Oxytocin drives uterine contractions ("afterpains"), which are more intense and prolonged after surgical delivery compared to vaginal birth
- The uterine incision repair is a potential nidus for endometritis if contaminated
Cardiovascular/VTE Physiology
- Elevated clotting factors (I, II, VII, VIII, X, XII), decreased protein S
- Reduced fibrinolysis
- Venous stasis from uterine compression of the iliac veins and IVC throughout pregnancy (not fully resolved immediately post-delivery)
- Immobility after surgery compounds this
- This triple hit (Virchow's triad: hypercoagulability + venous stasis + endothelial injury from surgery) explains why DVT occurs in 1-2% of cesarean patients and pulmonary embolism is a leading cause of maternal death
Gastrointestinal Pathophysiology (Post-Op Ileus)
- Neurogenic: surgical manipulation activates sympathetic pathways (mesenteric adrenergic reflexes) that inhibit propulsive peristalsis; the enteric nervous system is directly disturbed by intraperitoneal handling
- Inflammatory: peritoneal exposure releases prostaglandins and inflammatory mediators that suppress GI motility
- Pharmacological: opioid analgesics bind mu-receptors in the gut, blocking acetylcholine release and reducing contractility
- Electrolyte disturbances: hypokalemia (common post-op) reduces smooth muscle contractility
- The result: delayed passage of flatus, abdominal distension, bloating, nausea, inability to tolerate oral intake
1. Diet and Nutritional Management
Pathophysiology of Post-Op Fasting
- Prolonging the catabolic state
- Causing mucosal atrophy of the gut lining, impairing the mucosal immune barrier
- Delaying first lactation and breastmilk production
- Contributing to ileus (the absence of luminal contents reduces GI motility signalling)
Current Evidence-Based Approach (ERAS Society 2025 Update - AJOG)
| Phase | Recommendation | Rationale |
|---|---|---|
| Immediately post-op (PACU) | Ice chips or sips of water as tolerated | Stimulates GI motility, prevents mucosal atrophy |
| Once alert, hemodynamically stable | Light diet (toast, crackers, clear fluids) | Early enteral feeding reduces ileus, shortens stay |
| Avoid prolonged fasting | No routine NPO after surgery | Fasting worsens catabolism and delays recovery |
- The ERAS systematic review (Pinho & Costa, 2024, 19,001 women) confirmed that ERAS protocols including early feeding reduced hospital stay by ~14 hours and opioid consumption significantly
Specific Dietary Guidance
- Start with ice chips/water, then clear fluids (broth, diluted juice, herbal tea)
- Advance to a light/soft diet once patient passes flatus or has a bowel movement and is nausea-free
- Avoid gas-forming foods: beans, lentils, carbonated drinks, cabbage (worsen post-op distension)
- Avoid constipating foods: white bread, bananas, processed foods (constipation strains the wound)
- High-protein diet: 1.2-2 g protein/kg/day for wound repair, immune function, and muscle preservation (Sabiston)
- 25-30 kcal/kg/day total energy
- Carbohydrates: primary energy source; adequate intake prevents protein catabolism for fuel
- Healthy fats: omega-3 over omega-6; reduce inflammatory response
- High fibre: fruits, vegetables, whole grains - prevents constipation without straining the incision
- Adequate fluids: 2-3 L/day minimum; breastfeeding adds ~500 ml/day extra requirement
- Vitamin A: epithelial cell proliferation, tissue repair
- Vitamin C: collagen synthesis (directly supports incision healing)
- Vitamin E: antioxidant, reduces oxidative damage to healing tissues
- Iron: address post-op anaemia (Hb <11 g/dL is diagnostic of maternal anaemia); iron deficiency worsens fatigue, immune function, and milk production
- Vitamin K: blood clotting factors II, VII, IX, X
2. Positioning and Posture Post-Operatively
Pathophysiology of Why Position Matters
- Supine position in the immediate post-op period can cause aortocaval compression by the uterus if it remains enlarged, reducing uteroplacental blood flow and maternal cardiac output
- Immobility causes dependent venous pooling in the lower limbs, compounding the pro-thrombotic state
- A recumbent position reduces respiratory reserve volume, predisposing to atelectasis (most common early post-op pulmonary complication) and pneumonia
Positioning Protocol
- Semi-recumbent position (30-45 degrees): head of bed elevated to reduce aspiration risk (especially while neuraxial block wearing off), improve respiratory mechanics, reduce atelectasis
- Left lateral tilt if needed: relieves residual uterine pressure on aorta and IVC in the early post-op period
- The urinary catheter allows supine positioning without discomfort; once removed, mobilisation is expected
- Assess motor function return after neuraxial anesthesia (leg strength, proprioception)
- If adequate: sit on the edge of the bed - this alone provides respiratory and circulatory benefit
- If motor block has not resolved at 6 hours, medical review is indicated (may indicate rare complication like spinal haematoma)
- Supported transfer from bed to chair
- Ambulate as tolerated; aim to walk 1-2 times in the hallway under supervision
- Compression stockings or pneumatic sequential compression devices applied when in bed
- Walk 3-4 times in the hallway, aiming for out of bed for ≥8 hours during this period
- Independent ambulation expected for most patients by 24-36 hours
Wound Support Posture
- When getting up, advise patient to splint the incision: hold a pillow firmly against the abdomen when coughing, sneezing, or rising from lying. This reduces shear stress on the wound edges
- Avoid heavy lifting (>5 kg) for 6 weeks - the fascial layer (not just skin) needs time to remodel; tensile strength does not reach 80% until approximately 6 weeks
3. Mobilisation - Why It Is Non-Negotiable
- GI: bowel motility depends partly on physical activity and gravity; immobility prolongs ileus, delays first flatus, worsens bloating
- Respiratory: reduces functional residual capacity, collapses basal alveoli, causes atelectasis; early walking improves pulmonary function
- VTE: venous stasis in immobile legs, combined with the hypercoagulable post-partum state, makes DVT a real threat; early walking is the single most important mechanical VTE prevention measure
4. Specific Complication Management with Pathophysiology
4a. Post-Op Anaemia
- Check Hb on day 1 post-op
- Oral iron (ferrous sulphate 200 mg TDS) if Hb 7-10 g/dL and haemodynamically stable
- IV iron (ferric carboxymaltose) for faster repletion or if oral not tolerated
- Blood transfusion if Hb <7 g/dL or symptomatic (tachycardia, dyspnoea, chest pain)
- Recheck Hb at 6-week postnatal check
4b. Pain Management (Etiology and Mechanism)
- Somatic pain: from the abdominal wall incision - A-delta and C-fibre nociceptors are activated by the incision and sutured tissue; peaks day 1-2, resolves over 2-3 weeks
- Visceral pain: from uterine contractions (afterpains) driven by oxytocin; prostaglandins sensitise uterine nociceptors
- Referred pain: shoulder-tip pain from diaphragmatic irritation by residual intraperitoneal blood or air
- Scheduled paracetamol 1 g QDS (baseline - do not give PRN)
- NSAID (ibuprofen 400 mg TDS or diclofenac 50 mg TDS) - inhibit prostaglandin synthesis, reduce uterine cramps and somatic pain - add after paracetamol, not instead of
- Intrathecal morphine (given intra-op, provides 12-24 hours post-op analgesia) - most effective single agent
- TAP block or QL block if no neuraxial morphine given
- Oral opioids (codeine or oxycodone) for breakthrough only - minimise due to constipation, sedation, neonatal transfer in breastmilk
- Avoid codeine and tramadol in breastfeeding mothers - can cause neonatal CNS depression via ultra-rapid metabolism to morphine
- Adjuncts: gentle heat to uterine area for afterpains, positioning with pillow support
4c. Constipation (Extremely Common Post-Op)
- Lactulose 15 ml BD: osmotic laxative, draws water into the colon, softens stool - safe in breastfeeding
- Senna (senokot): stimulant laxative - use if lactulose insufficient
- Encourage early ambulation - most effective non-pharmacological intervention
- Adequate fluid intake (>2 L/day)
- High fibre diet once tolerating solid food
- Avoid routine enemas (not evidence-based post-cesarean)
4d. Wound Infection (Surgical Site Infection / Endometritis)
- Wound infection (SSI): occurs in 3% with prophylactic antibiotics. Most (25-30%) are from Staphylococcus aureus skin flora - not from endometrial contamination. The warm, moist environment under an abdominal pannus (especially in obese women) creates ideal conditions for bacterial growth. Disruption of the wound's inflammatory-proliferative healing cascade allows bacteria to colonise and form biofilm
- Endometritis: polymicrobial (anaerobes, Group B Strep, E. coli, Gardnerella, Enterococcus). The uterine incision, combined with cervical opening (especially in laboured cesareans), allows ascending vaginal flora to colonise the decidua and myometrium
- Wound: increasing redness beyond 2 cm of incision margin, warmth, swelling, purulent discharge, wound edges separating, foul odour
- Endometritis: fever >38°C after day 1, uterine tenderness on palpation, malodorous lochia, heavy bleeding, malaise
- Wound swab for M,C&S before starting antibiotics
- Co-amoxiclav 625 mg TDS x 7 days (covers skin flora including MRSA-negative Staph, anaerobes)
- If penicillin-allergic: clarithromycin + metronidazole
- For endometritis: refer to hospital for IV antibiotics (clindamycin + gentamicin is gold standard)
- Wound dressing: clean and dry; cover for first 48 hours; leave open to air after that if intact
- Do not use antiseptic solutions (povidone-iodine, hydrogen peroxide) routinely on healing wounds - they damage granulating tissue
- Wound dehiscence: do not attempt primary closure in infected wounds - manage by secondary intention, daily saline dressings; refer to surgical team
4e. DVT / VTE Prophylaxis
- Hypercoagulability: elevated clotting factors, decreased anticoagulants, post-partum state
- Venous stasis: immobility post-op, prior IVC/iliac vein compression from gravid uterus
- Endothelial injury: surgical trauma to pelvic vessels
- Graduated compression stockings from admission through discharge
- Pneumatic sequential compression devices (PCDs) while in bed/immobile
- LMWH (enoxaparin 40 mg SC OD): start 4-6 hours post-op once haemostasis confirmed; continue for 10 days minimum, up to 6 weeks in high-risk patients (obesity, personal/family history of VTE, thrombophilia, immobility)
- UFH (unfractionated heparin) as alternative if renal impairment
4f. Urinary Catheter and UTI
- Remove catheter 6-12 hours post-op, once patient is walking
- Do not leave in longer than necessary
- Monitor first void after removal: if unable to void in 4-6 hours, perform in-out catheterisation (do not re-insert indwelling)
- Encourage fluid intake >2 L/day to promote bladder flushing
- If symptomatic UTI (dysuria, frequency, fever): MSU for culture, treat with trimethoprim 200 mg BD x 7 days or nitrofurantoin 100 mg BD x 7 days (avoid nitrofurantoin in near-term/early breastfeeding); adjust based on sensitivities
5. Breastfeeding Support
- The absence of the labour process (and associated oxytocin surge during vaginal delivery) means the lactation hormonal cascade is triggered later
- Anaesthesia effects, pain, and maternal fatigue delay first feeding attempts
- Neonatal respiratory morbidity after pre-labour cesarean may mean brief NICU admission, disrupting skin-to-skin contact
- Encourage skin-to-skin contact immediately post-op as soon as mother is stable (even in theatre for elective cesareans)
- First feed within 1 hour of birth when possible
- Positioning: the wound incision limits traditional cradle hold; advise:
- Football/rugby ball hold: baby under arm, facing the breast, feet pointing behind mother - no pressure on incision
- Side-lying position: both mother and baby lying on their side, facing each other - comfortable post-op, good for overnight feeds
- Laid-back/biological nurturing: mother reclined, baby prone on chest - gravity holds baby, no incision pressure
- Arrange lactation consultant review before discharge
- Most analgesics are safe for breastfeeding at standard doses; avoid codeine, tramadol, and high-dose meperidine
6. Psychological Care and Postnatal Mental Health
- "Baby blues" (days 3-5): transient tearfulness, irritability, anxiety - affects 50-80% of women; resolves spontaneously; reassure
- Postnatal depression (PND): persistent low mood, anhedonia, anxiety, poor bonding beyond 2 weeks; affects ~10-15%; screen with Edinburgh Postnatal Depression Scale (EPDS) at 6-week check
- EPDS score ≥13: significant PND - refer to GP/mental health team
- Manage with CBT, peer support groups; SSRIs (sertraline preferred - minimal breast milk transfer) for moderate-severe cases
- PTSD after emergency cesarean: 3-9% of women; intrusive memories of the procedure, avoidance, hyperarousal; refer for trauma-focused CBT
7. Discharge Criteria and Follow-Up (Primary Care)
- Haemodynamically stable; Hb acceptable
- Pain controlled on oral analgesia
- Tolerating solid diet and adequate fluids
- Passed urine independently after catheter removal
- Passed flatus or had bowel movement
- Wound intact, no signs of infection
- Breastfeeding established or support arranged
- LMWH prescription supplied if indicated
- Discharge medications: paracetamol, ibuprofen, laxative, iron (if anaemic), LMWH
- No driving for 6 weeks (unable to perform emergency stop safely; also medicolegal)
- No heavy lifting >5 kg for 6 weeks (wound remodelling)
- Pelvic floor exercises from day 1
- No penetrative sex for 6 weeks (risk of ascending infection to uterine scar)
- Return if: fever, wound separation, offensive lochia, heavy bleeding (soaking >1 pad/hour), leg swelling, breathing difficulty, chest pain
- Day 7-10: wound check, BP, postnatal mental health screen, anaemia review
- 6-week check: full postnatal review - EPDS, contraception, wound healing, return to activities, smear due, discuss future pregnancy preferences (VBAC eligibility)
- ERAS Society Guidelines for Postoperative Care in Cesarean Delivery, 2025 Update (Sultan et al., AJOG 2026) - ERAS Society 2025 guidelines
- Pinho & Costa (2024) - Systematic review and meta-analysis of ERAS in cesarean delivery (19,001 women; PMID 38042118)
- Sabiston Textbook of Surgery, 21e (Postoperative Nutritional Care; Postoperative Ileus)
- Creasy & Resnik's Maternal-Fetal Medicine (Complications of Cesarean Delivery)
- Barash, Cullen & Stoelting's Clinical Anesthesia, 9e (Post-cesarean analgesia)
- O&G Magazine (Wound Healing Phases - Haemostasis, Inflammation, Proliferation, Remodelling)
What about diet, postural after post-op and, um, other general management or specific management with the causology and pathophysiology in details for a caesarean section patient in a primary?
- Pathophysiology of the surgical stress response, wound healing phases, uterine involution, VTE mechanisms, and post-op ileus
- Diet management - early feeding rationale, phased oral intake, macro/micronutrient requirements, breastfeeding nutrition
- Positioning and posture - semi-recumbent positioning, mobilisation timeline (0-8h, 8-24h, 24-48h), wound splinting technique
- Specific complication management with aetiology and pathophysiology for: post-op anaemia, pain, constipation, wound infection/endometritis, DVT/VTE, urinary catheter/UTI
- Breastfeeding positions safe after cesarean incision
- Postnatal mental health (baby blues, PND, PTSD) with pathophysiology
- Discharge criteria and follow-up schedule
Teach me congenital heart disease with pathophysiology and clinical presentation and treatment and why we are doing so with all the various genetic diseases that may have cardiac manifestations
Congenital Heart Disease (CHD) - A Complete Teaching Guide
PART 1: FOUNDATIONS
What Is CHD?
Embryological Basis - Why Does CHD Occur?
- The straight cardiac tube loops rightward and segments into atria, ventricles, and outflow tracts
- The endocardial cushions form the AV valves and membranous septum
- Neural crest cells migrate into the conotruncal (outflow tract) region to help form the aortopulmonary septum
- The septum primum and secundum grow to partition the atria; the interventricular septum grows upward from the apex
- Prematurity
- Family history (first-degree relative with CHD)
- Maternal conditions: diabetes (increases TGA, VSD, coarctation), phenylketonuria, connective tissue disorders, obesity
- Teratogenic drugs: phenytoin (pulmonary stenosis, ASD, aortic stenosis), retinoic acid (conotruncal defects), lithium (Ebstein anomaly), thalidomide
- In utero infections: rubella (PDA, pulmonary artery stenosis, VSD - the classic TORCH cardiac teratogen), CMV, coxsackievirus
- Chromosomal/genetic disorders (covered in detail in Part 4)
- Folate deficiency (supplementation reduces CHD incidence)
- Assisted reproductive technology (IVF)
PART 2: THE MASTER CLASSIFICATION FRAMEWORK
CHD
├── LEFT-TO-RIGHT SHUNTS (Acyanotic - early)
│ ├── ASD
│ ├── VSD (most common)
│ ├── PDA
│ └── AVSD (endocardial cushion defect)
│
├── RIGHT-TO-LEFT SHUNTS (Cyanotic - blue babies)
│ ├── Tetralogy of Fallot (most common cyanotic CHD)
│ ├── Transposition of Great Arteries (TGA)
│ ├── Truncus Arteriosus
│ ├── Total Anomalous Pulmonary Venous Return (TAPVR)
│ └── Tricuspid Atresia
│
└── OBSTRUCTIVE LESIONS (No shunt initially)
├── Pulmonary Stenosis
├── Aortic Stenosis / Atresia
└── Coarctation of the Aorta
- Left side of the heart normally operates at higher pressure than the right
- Therefore, any communication initially shunts left → right
- Only obstruction on the right side (e.g., pulmonary stenosis in ToF) or pulmonary hypertension severe enough to flip the gradient causes right → left shunting and cyanosis
PART 3: INDIVIDUAL LESIONS - Pathophysiology, Clinical Features, Treatment
Diagram of Left-to-Right Shunts (ASD, VSD, PDA)
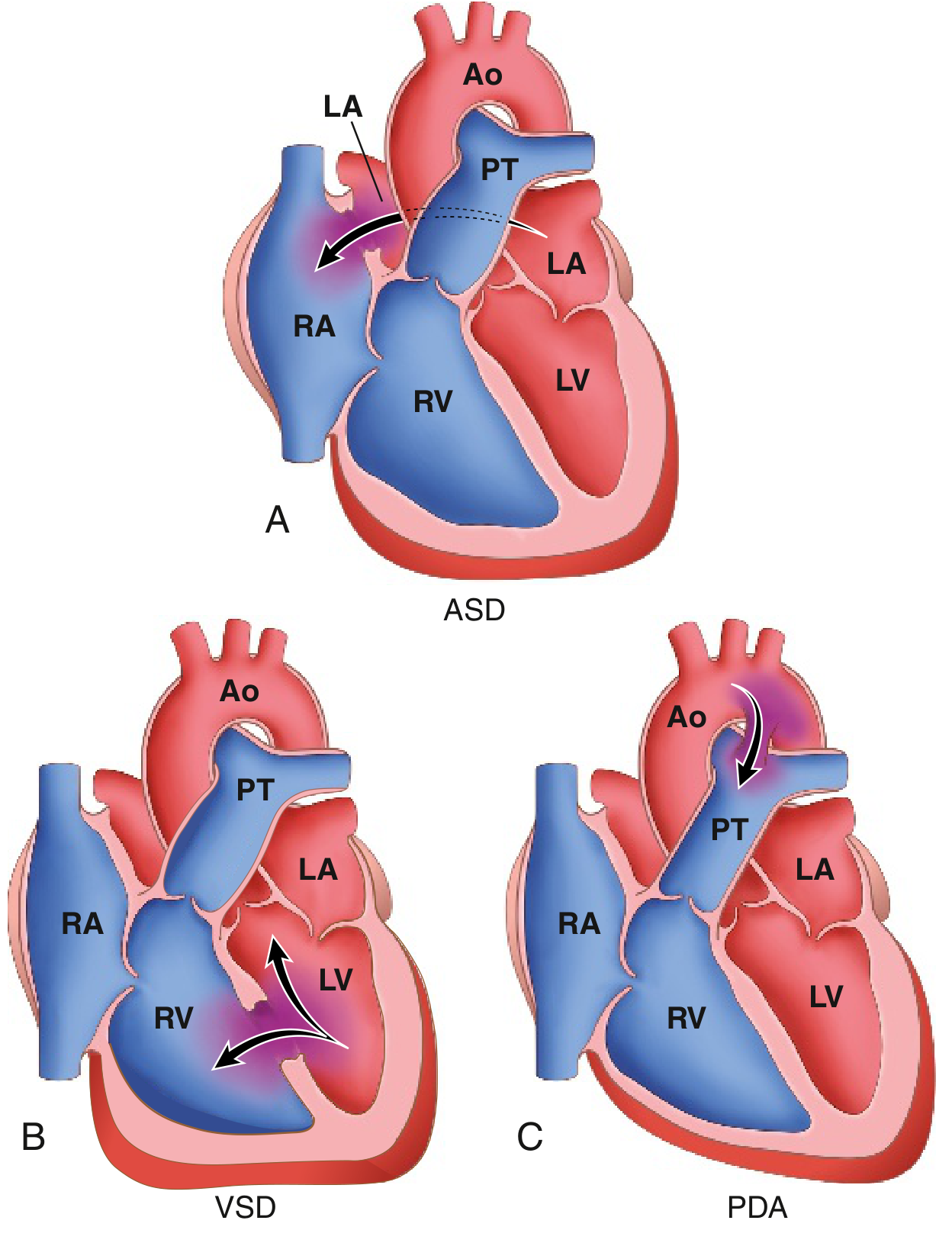
3.1 ATRIAL SEPTAL DEFECT (ASD)
Pathophysiology
- 90% are ostium secundum defects (near the fossa ovalis)
- 5% are ostium primum defects (at the lowest part - associated with AV valve abnormalities and Down syndrome)
- 5% are sinus venosus defects (high in septum, near SVC - associated with anomalous pulmonary venous drainage)
Clinical Presentation
- Often asymptomatic until adulthood (most commonly diagnosed CHD in adults)
- Exertional dyspnoea, fatigue, palpitations (AF/flutter from right atrial enlargement)
- Fixed wide splitting of S2 (delayed pulmonary valve closure due to persistent RV volume overload independent of respiration)
- Soft systolic ejection murmur at the upper left sternal border (increased flow across pulmonary valve - not the defect itself)
- Paradoxical embolism (DVT clot crosses ASD to systemic circulation → stroke)
- Late: right heart failure, cyanosis (Eisenmenger)
Treatment - Why and How
- Small defects (<1 cm): often close spontaneously; observe
- Percutaneous catheter closure (Amplatzer device): preferred for ostium secundum defects with adequate rim; low morbidity, outpatient procedure
- Surgical closure (direct suture or patch): for ostium primum, sinus venosus, and defects not amenable to device closure
- Timing: generally before school age or upon diagnosis in adults before Eisenmenger develops
- Eisenmenger syndrome: closure is contraindicated (would remove the "pressure release valve" for the right heart) - manage with pulmonary vasodilators (sildenafil, bosentan, prostacyclins)
- Antibiotic endocarditis prophylaxis: only for 6 months post-repair (or lifelong if residual defect)
3.2 VENTRICULAR SEPTAL DEFECT (VSD)
Pathophysiology
- Small VSD ("maladie de Roger"): small L→R shunt, limited haemodynamic significance; a small hole creates a high-velocity jet → loud murmur but little consequence
- Large VSD: large L→R shunt → increased pulmonary blood flow AND pressure → biventricular volume overload + pulmonary hypertension → biventricular failure
- Progressive pulmonary hypertension → Eisenmenger syndrome (as with ASD)
Clinical Presentation
- Small VSD: harsh pansystolic murmur at lower left sternal border (loud because of high-velocity jet through small orifice); often asymptomatic
- Large VSD: heart failure in infancy (poor feeding, failure to thrive, tachypnoea, sweating while feeding), pansystolic murmur may be softer (equal pressures = low velocity jet), mid-diastolic rumble (increased flow across mitral valve), hepatomegaly
- Recurrent lower respiratory tract infections (increased pulmonary blood flow)
- Late: Eisenmenger - paradoxically, VSD murmur gets quieter as pressures equalise, then right heart failure and cyanosis emerge
Treatment - Why and How
- 50% of small muscular VSDs close spontaneously in infancy/childhood - can observe
- Perimembranous and large VSDs: surgical patch repair or percutaneous device closure
- Timing: before irreversible pulmonary hypertension (before Eisenmenger)
- Heart failure management pre-repair: diuretics (furosemide), ACE inhibitors (reduce afterload, decrease shunt), adequate nutrition (high calorie feeds for failure to thrive)
- Infants with failure to thrive and uncontrolled heart failure: surgery in first months of life
- Pulmonary artery banding: older palliative technique - narrows PA surgically to reduce pulmonary blood flow until definitive repair possible
3.3 PATENT DUCTUS ARTERIOSUS (PDA)
Pathophysiology
- Increased oxygen tension with first breath (oxygen constricts the ductus smooth muscle)
- Increased bradykinin, decreased prostaglandin E2 (as placenta is removed)
Clinical Presentation
- Premature neonate: respiratory distress, inability to wean from ventilator, wide pulse pressure, bounding pulses, active precordium
- Full-term child/adult: continuous "machinery" murmur (because the shunt exists in both systole AND diastole), maximal at left infraclavicular region
- Wide pulse pressure (diastolic "run-off" into pulmonary circulation)
- Bounding peripheral pulses, collapsing pulse (Corrigan's pulse)
- In large PDA: failure to thrive, recurrent chest infections, eventually pulmonary hypertension
Treatment - Why and How
- Premature neonates: indomethacin IV or ibuprofen (COX inhibitors → reduce prostaglandin E2 → promote ductal closure); effective in ~70-80% of cases
- Surgical ligation: if pharmacological treatment fails or contraindicated (renal impairment, thrombocytopenia, NEC)
- Percutaneous catheter coil/device occlusion: preferred in older children and adults; Amplatzer duct occluder or coil embolisation
- Prostaglandin E1 (alprostadil): paradoxically used to KEEP the ductus OPEN in duct-dependent lesions (e.g., pulmonary atresia, critical coarctation) where PDA is the only source of pulmonary or systemic blood flow - this is a life-saving intervention in neonates
3.4 ATRIOVENTRICULAR SEPTAL DEFECT (AVSD) / Endocardial Cushion Defect
Pathophysiology
- Partial AVSD: ostium primum ASD + cleft mitral valve
- Complete AVSD: ostium primum ASD + inlet VSD + common AV valve (single valve between all four chambers)
Clinical Presentation
- Severe heart failure in infancy: poor feeding, failure to thrive, tachypnoea, diaphoresis, recurrent pneumonia
- Pansystolic murmur (VSD component), mid-diastolic flow murmur
- Hepatomegaly, crepitations
- CXR: cardiomegaly, pulmonary plethora
- ECG: superior axis deviation (pathognomonic - the AV node and His bundle are displaced superiorly by the absent lower atrial septum, resulting in leftward initial forces)
Treatment
- Surgical repair in first 3-6 months: patch closure of both ASD and VSD, reconstruction of the common AV valve into two separate valves
- Heart failure management pre-op: furosemide, ACE inhibitors, high-calorie feeds
- Down syndrome patients do as well post-operatively as chromosomally normal patients - Down syndrome is not a contraindication to surgery
3.5 TETRALOGY OF FALLOT (ToF)
Pathophysiology
- VSD (large, perimembranous - the displaced septum leaves a hole)
- Pulmonary outflow tract obstruction (usually subpulmonic stenosis - the displaced septum narrows the RV outflow)
- Overriding aorta (the aortic root straddles both ventricles, sitting over the VSD)
- Right ventricular hypertrophy (consequence of the outflow obstruction and RV pressure overload)
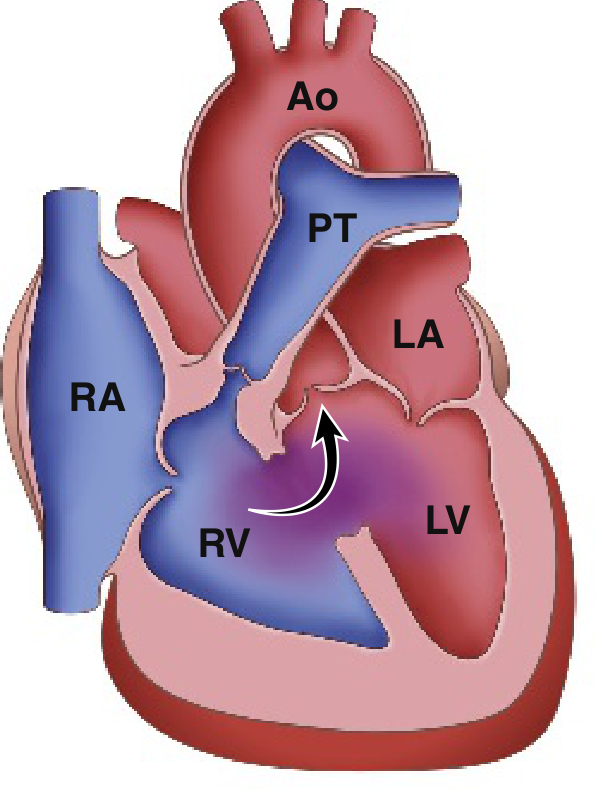
- Mild stenosis: still L→R shunting ("pink Tet") - resembles isolated VSD
- Severe stenosis: RV pressure exceeds LV pressure → right-to-left shunting through the VSD → deoxygenated blood enters the aorta → cyanosis
- Polycythaemia: hypoxia drives EPO production → increased RBC mass; helps O2 delivery but increases blood viscosity → thrombotic risk
- Clubbing: chronic hypoxia → periosteal vascular proliferation
- Hypertrophic osteoarthropathy
- Infective endocarditis risk (turbulent flow across VSD/pulmonary valve)
- Paradoxical embolism (venous thrombus crosses R→L into systemic circulation → stroke)
- Brain abscess: bacteria bypass pulmonary filter and seed the brain
Clinical Presentation
- Cyanosis from birth or early infancy (depending on severity)
- Harsh ejection systolic murmur at upper left sternal border (from pulmonary stenosis - NOT the VSD, since equal pressures mean no jet)
- Single loud S2 (pulmonary component absent or very soft)
- Boot-shaped heart on CXR (RV hypertrophy causes upturned apex; pulmonary bay concave due to hypoplastic PA)
- Right ventricular hypertrophy on ECG (right axis deviation, RV strain)
- Clubbing, polycythaemia in older unrepaired patients
- "Tet spells": acute cyanosis, hyperpnoea, loss of murmur
Treatment - Why and How
- Knee-chest position (increases SVR → reduces R→L shunt)
- Oxygen (though limited benefit if severe cyanosis)
- IV morphine (sedates, relieves infundibular spasm)
- IV propranolol (beta-blocker → relaxes infundibular muscle spasm)
- IV phenylephrine (alpha-agonist → increases SVR → reduces R→L shunt)
- IV fluid bolus (increases preload → maintains output)
- VSD patch closure
- RV outflow tract relief (resection of infundibular muscle, pulmonary valvotomy/valve reconstruction, transannular patch if necessary)
- Why repair early? Progressive infundibular hypertrophy worsens obstruction over time; the longer the right ventricle works against obstruction, the more irreversible the hypertrophy and fibrosis
- Blalock-Taussig-Thomas (BTT) shunt: subclavian artery to pulmonary artery shunt to increase pulmonary blood flow in very sick neonates unfit for full repair
- Prostaglandin E1 (alprostadil): keeps PDA open in neonates with pulmonary atresia/severe ToF, acting as a bridge to surgery
3.6 TRANSPOSITION OF THE GREAT ARTERIES (TGA)
Pathophysiology
- Right heart → deoxygenated blood → aorta → body → right heart (closed loop)
- Left heart → oxygenated blood → pulmonary artery → lungs → left heart (closed loop)
- PDA (common initially, but closes in first days)
- Patent foramen ovale / ASD
- VSD (in ~1/3 of cases)
Clinical Presentation
- Severe cyanosis within first hours of life (as PDA closes)
- Cyanosis paradoxically NOT improved by 100% oxygen (hyperoxia test - oxygen saturation remains low because the lungs are connected to the wrong ventricle)
- Tachypnoea but without severe respiratory distress early (lungs not flooded)
- CXR: "egg-on-string" appearance (narrow superior mediastinum - the aorta and PA are parallel not crossed; large globular heart)
- Without intervention: death within days to weeks
Treatment
- Emergency balloon atrial septostomy (Rashkind procedure): catheter inserted via umbilical vein, balloon inflated in left atrium and dragged across the foramen ovale, tearing it open to create an ASD → allows mixing → buys time
- Prostaglandin E1: keeps PDA open to provide mixing
- Definitive: Arterial Switch Operation (Jatene procedure) within the first 1-2 weeks of life:
- The aorta and PA are divided and switched to the correct ventricles
- The coronary arteries must be reimplanted into the neo-aorta (technically demanding)
- Why so urgent? After birth, the left ventricle pumps only to the low-resistance lungs; within 2-4 weeks it becomes too "de-trained" (too thin-walled) to handle systemic pressures after the switch
- Outcomes excellent with modern surgery (>95% survival)
3.7 COARCTATION OF THE AORTA
Pathophysiology
- Preductal ("infantile"): diffuse hypoplasia of the aortic arch proximal to a patent ductus - presents in neonates when the ductus closes; lower body perfusion was dependent on right-to-left PDA flow
- Postductal ("adult"): discrete shelf-like infolding of the aorta distal to the ligamentum arteriosum; collateral circulation develops through intercostal arteries
- Obstruction to left ventricular outflow → LV hypertension → LV hypertrophy → LV failure
- Hypertension in the upper body (arms, head): because the aorta proximal to the coarctation is under high pressure
- Hypotension and hypoperfusion in the lower body (legs, kidneys): because the aorta distal to the coarctation is under low pressure
- Collateral circulation develops (intercostal arteries enlarge) → "rib notching" on CXR
- Bicuspid aortic valve is present in 50% of coarctation cases (both are left-sided obstructive lesions, sharing genetic aetiology - haploinsufficiency of NOTCH1 or other left-sided cardiac genes)
- Circle of Willis aneurysms associated (Berry aneurysms) → risk of subarachnoid haemorrhage
Clinical Presentation
- Shock when the PDA closes (lower body was getting blood via R→L PDA flow)
- Differential cyanosis: upper body pink (pre-ductal blood), lower body cyanosed (post-ductal blood from RV via PDA)
- Absent or weak femoral pulses, diminished BP in legs
- Upper limb hypertension with weak/absent femoral pulses - key clinical sign
- Radio-femoral delay (femoral pulse arrives after radial pulse)
- BP differential >20 mmHg between arms and legs
- Headache, epistaxis (from hypertension)
- Leg claudication on exercise (lower body underperfused)
- Systolic murmur over the chest/back (at the coarctation site) and over collateral vessels (intercostal arteries)
- CXR: "Figure of 3" sign (indentation of the aorta at coarctation site), rib notching (inferior surface, ribs 3-8, from collateral intercostal vessels)
Treatment - Why and How
- Neonatal emergency: prostaglandin E1 to maintain ductal patency → stabilise → surgical repair
- Surgical options: resection and end-to-end anastomosis (preferred in neonates/infants), subclavian flap aortoplasty, patch aortoplasty, bypass graft
- Balloon angioplasty ± stenting: preferred for older children, adolescents, and adults; lower morbidity than surgery; stenting reduces re-coarctation rate
- Treat bicuspid aortic valve if significant stenosis/regurgitation
- Life-long monitoring: even after repair, residual hypertension, re-coarctation, and aortic aneurysm at repair site remain risks; annual echo and BP monitoring in both arms and legs
- Berry aneurysm: neurosurgical or endovascular treatment if symptomatic
3.8 EISENMENGER SYNDROME (End-Stage Shunt Disease)
- Initial L→R shunt → increased pulmonary blood flow and pressure
- Pulmonary arteriolar smooth muscle hypertrophy (medial thickening) - reversible
- Intimal fibrosis, plexiform lesions - irreversible
- Pulmonary vascular resistance (PVR) rises progressively
- When PVR ≥ systemic vascular resistance (SVR): shunt reverses (R→L) → cyanosis
- Pulmonary vasodilators: sildenafil (PDE5 inhibitor), bosentan (endothelin receptor antagonist), prostacyclins (epoprostenol, iloprost) - reduce PVR, improve symptoms and exercise tolerance
- Avoid: dehydration, exertion, high altitude, systemic vasodilators (reduce SVR → worsen R→L shunt)
- Anticoagulation: controversial (risk of haemoptysis vs. thromboembolic risk)
- Iron supplementation (secondary polycythaemia causes iron depletion)
- Heart-lung transplant: only definitive option; rare due to organ shortage
- Pregnancy is absolutely contraindicated in Eisenmenger (maternal mortality 30-50%)
PART 4: GENETIC SYNDROMES WITH CARDIAC MANIFESTATIONS
Master Table: Genetic Syndromes and Their Cardiac Lesions
| Syndrome | Genetic Defect | Cardiac Lesion(s) | Key Non-Cardiac Features |
|---|---|---|---|
| Down (Trisomy 21) | Trisomy 21 (extra chr 21) | AVSD (40%), VSD, ASD | Intellectual disability, flat facies, single palmar crease, Brushfield spots, Hirschsprung's, duodenal atresia, leukaemia |
| Turner (45,X) | Monosomy X | Coarctation (30%), bicuspid aortic valve, aortic dilation | Short stature, webbed neck, wide-spaced nipples, streak ovaries, lymphoedema, infertility |
| DiGeorge (22q11.2 del) | TBX1 deletion on chr 22q11 | Conotruncal defects (75%): truncus arteriosus, interrupted aortic arch, ToF, VSD, TGA | Thymic hypoplasia (T-cell deficiency), hypoparathyroidism (hypocalcaemia), palatal defects (velopharyngeal insufficiency), learning difficulties; also called CATCH-22 or velocardiofacial syndrome |
| Noonan | PTPN11, SOS1, KRAS (Ras-MAPK pathway) | Pulmonary valve stenosis (50%), AVSD, hypertrophic cardiomyopathy, ASD | Short stature, low-set ears, hypertelorism, ptosis, webbed neck (similar to Turner but normal karyotype), cryptorchidism |
| Williams | ELN deletion chr 7q11 | Supravalvular aortic stenosis (70%), peripheral pulmonary artery stenosis | "Elfin" facies, cognitive delay with exceptional verbal/musical ability, hypercalcaemia, friendly personality |
| Holt-Oram | TBX5 (transcription factor) | ASD, VSD, conduction defects (AV block) | Radial ray limb defects (absent thumb/radius), "heart-hand" syndrome |
| Marfan | FBN1 (fibrillin-1, chr 15) | Aortic root dilation, aortic dissection, mitral valve prolapse | Tall stature, arachnodactyly, arm span > height, lens dislocation (ectopia lentis), high-arched palate, pectus deformity |
| Loeys-Dietz | TGFBR1/2 mutations | Aortic root dilation (higher rupture risk than Marfan at smaller diameters), arterial tortuosity | Hypertelorism, bifid uvula, craniosynostosis; aortic dissection at younger age and smaller size than Marfan |
| Ehlers-Danlos (vascular type) | COL3A1 (type III collagen) | Aortic/arterial dissection and rupture | Thin translucent skin, easy bruising, spontaneous rupture of hollow organs (colon, uterus, arteries) |
| CHARGE syndrome | CHD7 (helicase-binding, chr 8) | ASD, VSD, PDA, hypoplastic right heart (75%) | Coloboma, Heart, choanal Atresia, Retarded growth, Genital abnormalities, Ear anomalies/deafness |
| Alagille | JAG1 or NOTCH2 (Notch signalling) | Peripheral pulmonary artery stenosis, ToF | Bile duct paucity (cholestasis), characteristic facies (broad forehead, pointed chin), butterfly vertebrae, posterior embryotoxon |
| Trisomy 18 (Edwards) | Trisomy 18 | VSD, ASD, PDA, polyvalvular disease | Clenched fists with overlapping fingers, rocker-bottom feet, choroid plexus cysts, severe intellectual disability; most die within first year |
| Trisomy 13 (Patau) | Trisomy 13 | VSD, ASD, PDA, dextrocardia | Holoprosencephaly, cyclopia, cleft lip/palate, polydactyly; most die within days to weeks |
| Char syndrome | TFAP2B | PDA | Characteristic facies, fifth finger clinodactyly |
| Friedreich's Ataxia | GAA repeat expansion (Frataxin gene) | Hypertrophic cardiomyopathy (95% of patients; primary cause of death) | Progressive spinocerebellar ataxia, loss of proprioception, diabetes mellitus |
| Pompe disease (GSD type II) | GAA enzyme deficiency (lysosomal storage) | Hypertrophic cardiomyopathy (massive - "glycogen-laden heart") | Generalised hypotonia, respiratory failure; infantile form fatal without enzyme replacement therapy |
| Fabry disease | Alpha-galactosidase A deficiency (X-linked) | Hypertrophic cardiomyopathy, conduction disease, valve disease | Neuropathic pain, angiokeratomas, renal failure |
| LEOPARD / Cardiofaciocutaneous syndrome | PTPN11, RAF1 (Ras-MAPK) | Hypertrophic cardiomyopathy, pulmonary stenosis | Lentigines, ECG abnormalities, Ocular hypertelorism, Pulmonary stenosis, Abnormal genitalia, Retardation of growth, Deafness |
In-Depth: The Most Clinically Important Genetic-Cardiac Connections
Down Syndrome (Trisomy 21) → AVSD
DiGeorge Syndrome (22q11.2 Deletion) → Conotruncal Defects
- The thymus (→ T-cell immunodeficiency)
- The parathyroid glands (→ hypocalcaemia)
- The conotruncal region of the heart (→ outflow tract defects)
- Cardiac defects (conotruncal)
- Abnormal facies
- Thymic hypoplasia
- Cleft palate
- Hypocalcaemia
- 22q11 deletion
Turner Syndrome (45,X) → Coarctation / Bicuspid Aortic Valve
Marfan Syndrome (FBN1) → Aortic Root Disease
- Progressive aortic root dilation (Z-score >2 is abnormal)
- Aortic regurgitation (dilated root distorts valve coaptation)
- Mitral valve prolapse (myxomatous degeneration) and mitral regurgitation
- Aortic dissection (type A - ascending) is the primary cause of death
- Beta-blockers (propranolol, atenolol): reduce dP/dT (rate of pressure rise in the aorta) → less mechanical stress on the aortic wall → slow dilation rate
- Losartan (ARB): blocks TGF-beta signalling → may reduce rate of aortic dilation independent of blood pressure lowering; used particularly in children
- Aortic root surgery when root diameter reaches 4.5-5 cm (or earlier if rapid progression): valve-sparing root replacement (David procedure) or Bentall procedure (composite valve-graft)
- Avoid: contact sports, competitive athletics, isometric exercise, pregnancy until aortic root is addressed; avoid fluoroquinolones (collagen-disrupting)
Noonan Syndrome → Pulmonary Stenosis / HCM
PART 5: TREATMENT PRINCIPLES - THE "WHY" FRAMEWORK
The Universal Principle: Intervene Before Irreversibility
| Stage | What is happening | What intervention achieves |
|---|---|---|
| L→R shunt + normal pulmonary pressures | Pulmonary vasculature still normal; RV dilated but healthy | Closure prevents pulmonary hypertension entirely |
| L→R shunt + elevated (but responsive) pulmonary pressures | Early medial hypertrophy; reversible with closure | Closure allows regression of pulmonary vascular changes |
| Eisenmenger (fixed pulmonary hypertension) | Plexiform lesions, intimal fibrosis - irreversible | Closure is now harmful (removes pressure relief); manage medically only |
Drug Classes Used in CHD
| Drug | Mechanism | When Used | Why |
|---|---|---|---|
| Prostaglandin E1 (alprostadil) | Keeps ductus arteriosus open | Duct-dependent lesions (pulmonary atresia, critical coarctation, TGA) | Maintains pulmonary or systemic blood flow until surgical correction |
| Indomethacin / Ibuprofen | COX inhibitor → reduces PGE2 → promotes ductal closure | PDA in premature neonates | Closes the ductus pharmacologically, avoiding surgery |
| Diuretics (furosemide) | Loop diuretic → reduces preload | Heart failure from L→R shunts (VSD, AVSD, PDA) | Reduces pulmonary oedema and congestion |
| ACE inhibitors | Reduce afterload and preload | Heart failure from L→R shunts | Reduce systemic vascular resistance → less L→R shunting; also protect kidneys and myocardium |
| Beta-blockers | Reduce heart rate, dP/dT, myocardial oxygen demand | Tet spells (propranolol), Marfan (slow aortic dilation), HCM | Reduce infundibular spasm in ToF; reduce shear stress on Marfan aorta |
| Phenylephrine (alpha-agonist) | Increases SVR | Tet spells | Raises aortic pressure → reduces R→L shunting through VSD |
| Morphine | Sedation, reduces peripheral vasodilation, relaxes infundibular spasm | Tet spells | Multiple mechanisms to reduce cyanosis crisis |
| Sildenafil | PDE5 inhibitor → ↑ cGMP → pulmonary vasodilation | Eisenmenger syndrome, pulmonary arterial hypertension | Selectively dilates pulmonary vessels, reducing PVR |
| Bosentan | Endothelin receptor antagonist | Eisenmenger syndrome | Endothelin is a potent pulmonary vasoconstrictor; blocking it reduces PVR |
| Losartan | ARB; also blocks TGF-beta signalling | Marfan syndrome | Slows aortic root dilation by reducing TGF-beta mediated matrix degradation |
Summary: CHD in One Mental Model
EMBRYOGENESIS DISRUPTED (wks 3-8)
↓
STRUCTURAL DEFECT
↓
┌─────────────────────────────────────────────┐
│ L→R SHUNT │ R→L SHUNT │ OBSTRUCTION │
│ (initially │ (cyanosis │ (pressure │
│ acyanotic) │ from birth) │ overload) │
│ ASD, VSD, PDA, │ ToF, TGA, │ CoAo, │
│ AVSD │ Truncus, │ PS, AS │
│ │ TAPVR │ │
└─────────────────────────────────────────────┘
↓ ↓ ↓
Pulmonary Systemic LV/RV
hypertension hypoxia failure
↓ ↓ ↓
Eisenmenger Polycythaemia, Surgical
syndrome clubbing, repair
↓ brain abscess
Irreversible →
no closure →
pulmonary vasodilators
- Robbins & Kumar Basic Pathology, 10e, Chapter 9 (Congenital Heart Disease - complete pathology basis)
- Robbins, Cotran & Kumar Pathologic Basis of Disease - Table 12.3 (Gene defects in CHD)
- Goldman-Cecil Medicine (Adult CHD, Tetralogy of Fallot in adults)
- Mulholland & Greenfield's Surgery, 7e (AVSD and Down syndrome)
- Harriet Lane Handbook, 23e (Genetic syndromes with cardiac defects - paediatric reference table)
- Emery's Elements of Medical Genetics (Noonan syndrome and RASopathies)
- Braunwald's Heart Disease (Marfan and inherited aortopathies)