Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome" AND pathophysiology
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have comprehensive textbook content and two excellent diagrams. I have enough to write a detailed, well-organized answer.
Acute Respiratory Distress Syndrome (ARDS): Mechanism
Overview
ARDS is a syndrome of acute diffuse inflammatory lung injury characterized by non-cardiogenic pulmonary edema caused by increased alveolar-capillary permeability. Unlike congestive heart failure (where hydrostatic pressure drives edema), in ARDS the edema fluid is exudative - protein-rich fluid leaks into the airspaces because the barrier itself is disrupted. The result is impaired gas exchange, decreased compliance, right-to-left shunting, and profound hypoxemia.
- Murray & Nadel's Textbook of Respiratory Medicine, p. 3069
Phases of ARDS
The timeline below illustrates the three overlapping phases:

Figure: Time course of ARDS phases. Harrison's Principles of Internal Medicine 22E
Phase 1: Exudative (Days 0-7)
This is the acute injury phase. Its defining pathology is diffuse alveolar damage (DAD).
The Alveolar-Capillary Barrier Fails
Two cellular layers normally form this barrier - the type I/II alveolar epithelium (airspace side) and the microvascular endothelium (blood side). In ARDS, both are damaged:
-
Epithelial injury is considered the key precipitating event. Type I pneumocytes (which cover ~95% of the alveolar surface) die via necrosis and apoptosis, disrupting the barrier and eliminating the cells responsible for active fluid clearance (via Na+/K+-ATPase and ENaC channels). Type II pneumocytes, which produce surfactant and can differentiate into type I cells, are also injured.
-
Endothelial injury increases microvascular permeability, which is both necessary and sufficient for edema formation. Tight junctions between endothelial cells are disrupted, allowing protein-rich fluid to flood the interstitium and alveoli.
-
Murray & Nadel's Textbook of Respiratory Medicine, p. 3083
The Central Role of Neutrophils
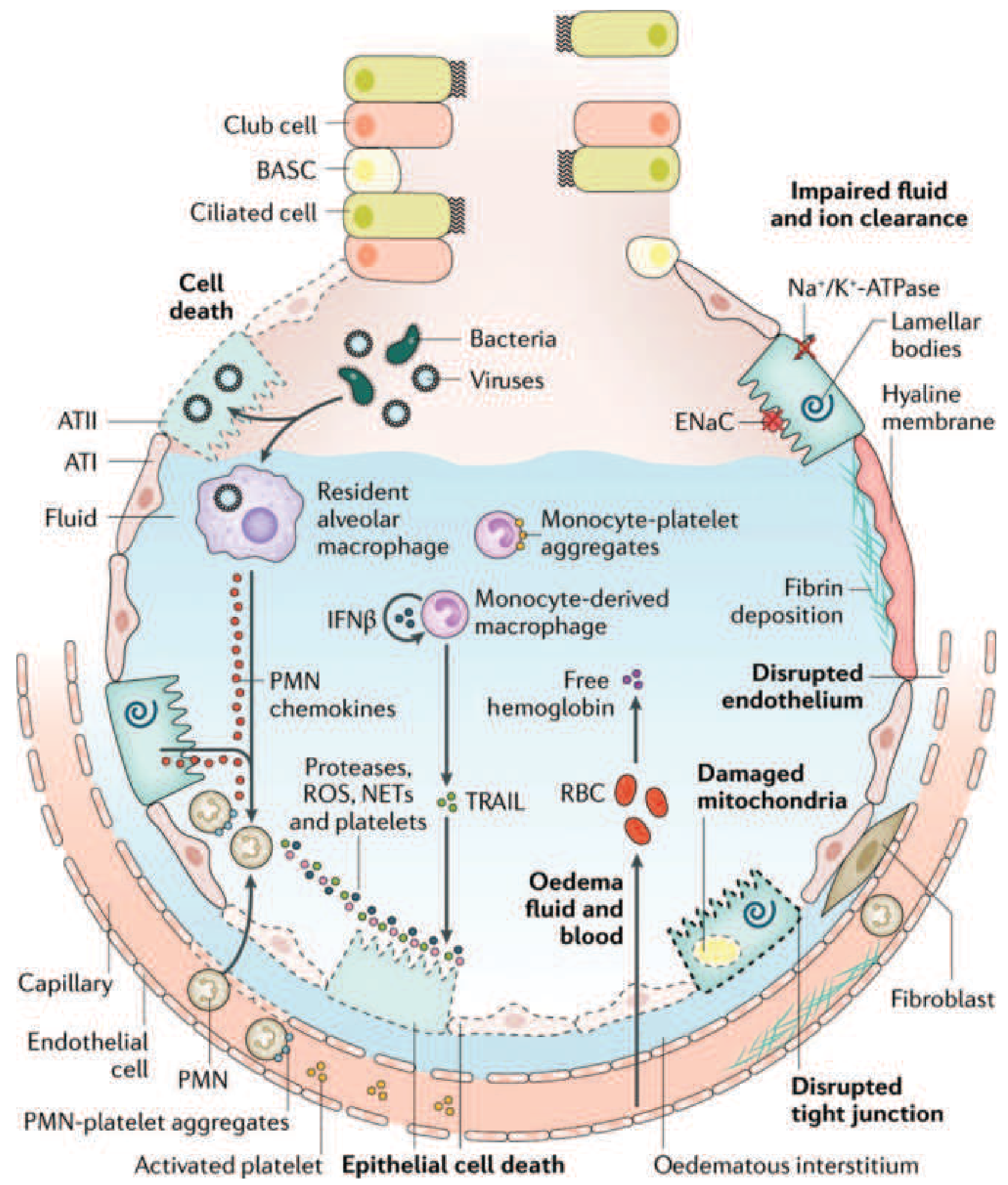
Figure 312-3: The injured alveolus in the acute phase of ARDS. Harrison's Principles of Internal Medicine 22E
Neutrophils are the principal effector cells of acute lung injury. The sequence of events:
- Sequestration: One of the earliest signs of ARDS (even before hypoxemia) is a transient peripheral leukopenia as neutrophils are sequestered in the pulmonary microvasculature. Pulmonary capillaries (~5 µm) are narrower than neutrophils (~7-8 µm), so neutrophils must deform to pass through. Activated neutrophils become "stiff" (via actin cytoskeleton changes) and cannot deform, causing them to accumulate.
- Transmigration: Sequestered neutrophils migrate across the alveolar-capillary membrane into the interstitium and airspaces. Importantly, in the lung this can occur without the adhesion molecules (L-selectin, β2-integrins) typically required elsewhere.
- Cytotoxic payload release: Once in the alveolar space, activated neutrophils release:
- Reactive oxygen species (ROS) - cause oxidative damage to lipids, proteins, and DNA
- Neutrophil elastase (NE) - a serine protease that degrades epithelial and endothelial cadherins (components of adherens junctions), directly predisposing to alveolar flooding; also degrades surfactant proteins
- Matrix metalloproteinases - degrade extracellular matrix
- Neutrophil extracellular traps (NETs) - webs of chromatin and proteases that cause tissue damage
- Cytokines (TNF-α, IL-1β, IL-8) that amplify the inflammatory cascade
- Murray & Nadel's Textbook of Respiratory Medicine, pp. 3111-3151
Macrophage and Cytokine Activation
Resident alveolar macrophages sense the initial insult (bacteria, viruses, DAMPs) via Toll-like receptors and produce a cascade of pro-inflammatory mediators:
- TNF-α and IL-1β act as "master cytokines," activating endothelial cells to upregulate adhesion molecules (ICAM-1, E-selectin), driving further neutrophil recruitment
- IL-8 (CXCL8) is the principal neutrophil chemoattractant - markedly elevated in BAL fluid of ARDS patients
- Platelet-activating factor (PAF) enhances vascular permeability and activates platelets
- Monocyte-derived macrophages are recruited from circulation and sustain the inflammatory response
- p38 MAP kinase and phosphatidylinositol 3-kinase pathways are central intracellular signaling nodes that drive these cytokine responses
Surfactant Dysfunction
Normal surfactant (dipalmitoylphosphatidylcholine/DPPC) reduces alveolar surface tension and prevents collapse. In ARDS:
-
Surfactant production by type II pneumocytes is reduced
-
Neutrophil elastase degrades surfactant proteins A, B, and C
-
Plasma proteins that leak into the alveolar space inhibit surfactant function
-
The ratio of large (active) to small (inactive) surfactant aggregates falls
-
Net result: alveolar instability, widespread microatelectasis, and reduced compliance
-
Murray & Nadel's Textbook of Respiratory Medicine, pp. 3092-3101
Coagulation Abnormalities
ARDS is associated with a pro-coagulant, anti-fibrinolytic state in the alveolar compartment:
- Tissue factor is expressed on damaged epithelial cells and activated macrophages, triggering extrinsic coagulation
- Fibrin deposition occurs in alveolar spaces and within pulmonary microvessels (contributing to the hyaline membranes seen histologically and to microvascular occlusion)
- Plasminogen activator inhibitor-1 (PAI-1) levels are markedly elevated, suppressing fibrinolysis
Pulmonary Hypertension
Multiple mechanisms drive elevated pulmonary artery pressure:
- Hypoxic vasoconstriction
- Intravascular fibrin and platelet microthrombi occlude vessels
- Compression of blood vessels by positive-pressure ventilation
- Progressive vascular remodeling in prolonged disease
Phase 2: Proliferative (Days 7-21)
In patients who survive the exudative phase, a reparative process begins:
- Neutrophil-predominant infiltrate shifts to a lymphocyte-predominant pattern
- Hyaline membranes are reorganized and cleared
- Type II pneumocytes proliferate along alveolar basement membranes, synthesizing new surfactant and differentiating into type I cells to restore the epithelial barrier
- Alveolar exudates begin to organize (organizing pneumonia pattern)
- Early interstitial and alveolar fibrosis may appear in some patients
Most patients begin to recover clinically during this phase, though dyspnea, tachypnea, and hypoxemia often persist.
- Harrison's Principles of Internal Medicine 22E, p. 2688
Phase 3: Fibrotic (>21 Days)
A subset of patients (particularly those who remain on mechanical ventilation >2 weeks) enters a fibrotic phase:
-
Alveolar edema and exudates are replaced by extensive alveolar-duct and interstitial fibrosis
-
Disrupted acinar architecture creates emphysema-like changes with large bullae
-
Intimal fibroproliferation in the pulmonary microcirculation causes progressive vascular occlusion and worsening pulmonary hypertension
-
Physiologic consequences: reduced lung compliance, increased dead space, increased pneumothorax risk
-
Elevated N-terminal procollagen peptide III in BAL fluid (a marker of collagen synthesis) can be detected as early as 24 hours after onset, indicating fibrogenesis begins very early
-
Harrison's Principles of Internal Medicine 22E, p. 2690; Murray & Nadel's, p. 3074
Pathophysiologic Summary Table
| Feature | Mechanism | Consequence |
|---|---|---|
| Barrier breakdown | Epithelial/endothelial injury | Protein-rich alveolar edema |
| Neutrophil infiltration | Sequestration → transmigration → activation | ROS, elastase, NET-mediated tissue damage |
| Cytokine storm | Macrophage/monocyte activation (TNF-α, IL-1β, IL-8) | Amplified inflammation, more neutrophil recruitment |
| Surfactant dysfunction | NE degradation + plasma protein inhibition | Microatelectasis, reduced compliance |
| Fibrin deposition | Pro-coagulant state, PAI-1 elevation | Hyaline membranes, microvascular occlusion |
| Impaired fluid clearance | Loss of Na+/K+-ATPase on type I pneumocytes | Edema persists despite resolution of leak |
| Pulmonary hypertension | Hypoxic vasoconstriction + microvascular occlusion | Right heart strain |
Pathologic Definition: Diffuse Alveolar Damage (DAD)
DAD is the histologic correlate of ARDS, but importantly only about 50% of ARDS patients show DAD on autopsy or biopsy. Patients with confirmed DAD are younger, more severely ill, have lower PaO2/FiO2 ratios, require higher driving pressures, and are five times more likely to die of hypoxemic respiratory failure. The remainder have other pathologies (pneumonia, hemorrhage, eosinophilic pneumonitis) that mimic ARDS clinically.
- Murray & Nadel's Textbook of Respiratory Medicine, p. 3078
Recent Evidence
Two recent reviews (2024-2025) are consistent with this framework and note evolving areas:
- Wick KD et al., BMJ 2024 (PMID 39467606) - comprehensive review confirming neutrophil-platelet interactions, NETs, and the importance of ARDS phenotyping (hyperinflammatory vs. hypo-inflammatory subtypes) as an emerging mechanistic/therapeutic distinction
- Al-Husinat L et al., Critical Care 2025 (PMID 39994815) - highlights the 2023 Global definition update and emphasizes evolving understanding of ARDS subphenotypes and tailored management based on biological mechanisms
This is a shared conversation. Sign in to Orris to start your own chat.