Malignant ewings sarcoma histopathology description and dd
Reading File
Finding Sources
Searching PubMed
"Ewing sarcoma" histopathology diagnosis
Reading File
Reading File
Loading Image
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have comprehensive information from multiple authoritative sources. Here is the full, detailed answer:
Ewing Sarcoma - Histopathology & Differential Diagnosis
Overview
Ewing sarcoma is the second most common primary malignant bone tumor in patients under 20 years (after osteosarcoma) and the most common in children under 10. It accounts for ~10-15% of all primary malignant bone tumors, with approximately 200 new cases per year in the US. There is a slight male predominance and it is exceedingly rare in individuals of African descent.
Histopathology
Gross Appearance
- Usually diaphyseal, arising in the medullary cavity
- Invades cortex, periosteum, and soft tissue
- Soft, tan-white tumor with frequent hemorrhage and necrosis
- Radiologically shows destructive lytic lesion with permeative margins and characteristic "onion-skin" periosteal reaction (layers of reactive bone); a "sunburst" pattern may also be seen
Microscopic Appearance
FIG. 19.23 (Robbins Basic Pathology) - Ewing sarcoma: sheets of small round cells with minimal clear cytoplasm:
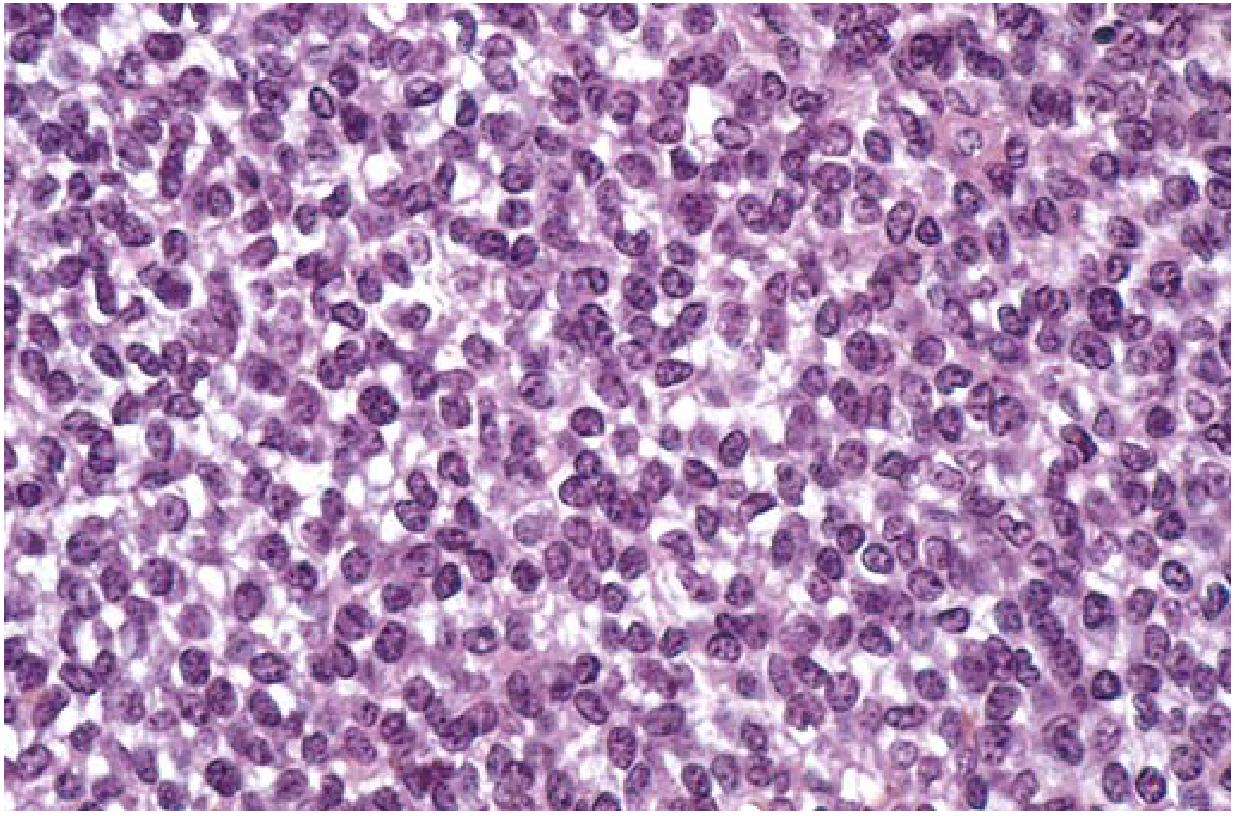
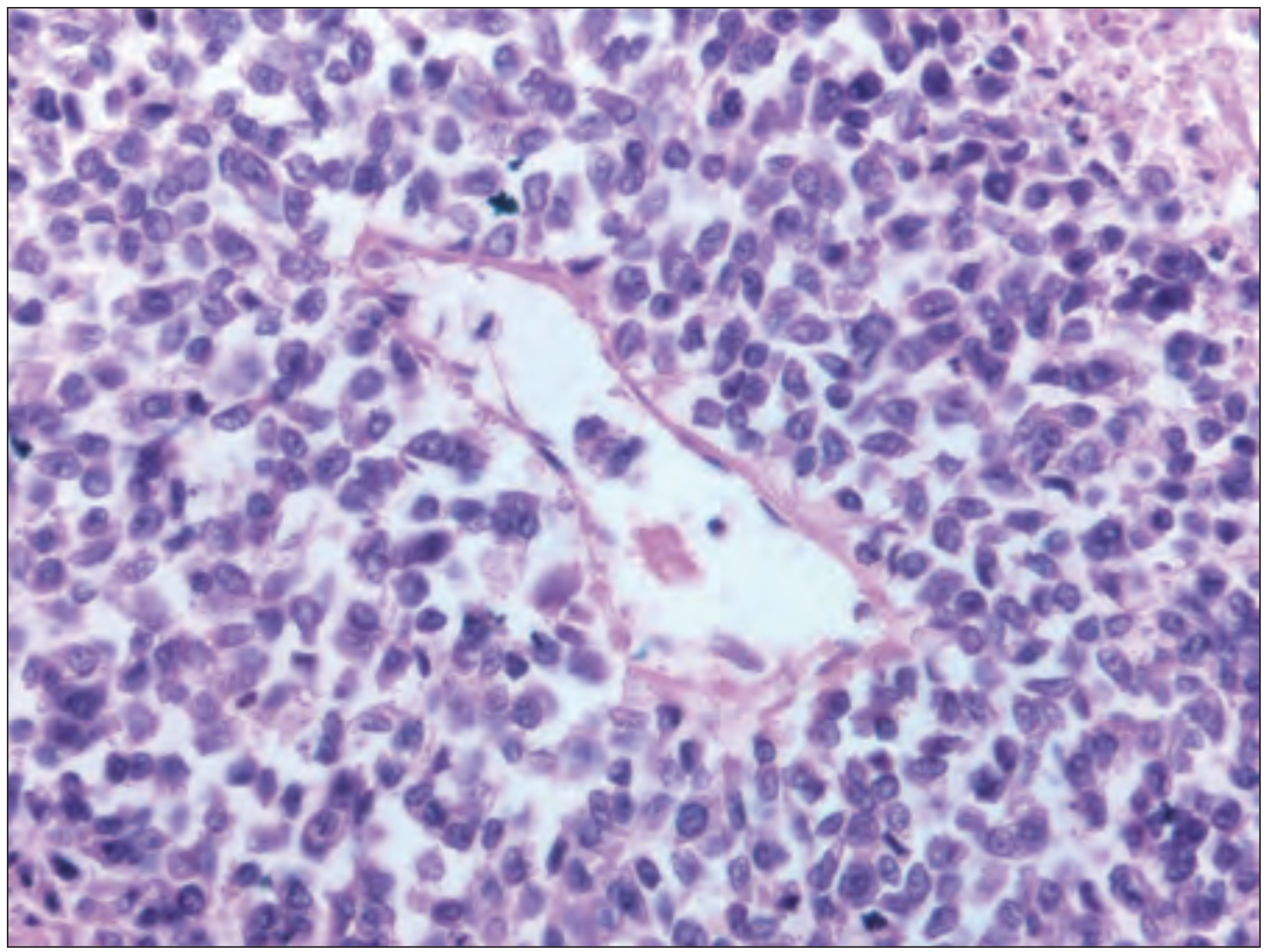
FIG. E218.38 (Rheumatology, Elsevier) - 60x photomicrograph showing small round blue cell tumor consistent with Ewing sarcoma, with prominent vascular spaces:
Key Microscopic Features:
| Feature | Description |
|---|---|
| Cell type | Uniform small round cells, slightly larger and more cohesive than lymphocytes |
| Cytoplasm | Scant; often appears clear (rich in glycogen - PAS positive, diastase sensitive) |
| Nuclei | Round, fine chromatin ("salt and pepper") |
| Nucleoli | Usually inconspicuous |
| Architecture | Broad sheets / diffuse pattern |
| Homer-Wright rosettes | May be present (circular groupings with central fibrillary core) - indicates neuroectodermal differentiation |
| Matrix | No bone or cartilage production (key negative feature) |
| Stroma | Scant fibrovascular stroma |
| Mitoses/Necrosis | Necrosis common; mitoses variable |
Immunohistochemistry (IHC)
| Marker | Ewing Sarcoma |
|---|---|
| CD99 (MIC2) | Strongly positive (diffuse membranous) - most characteristic marker |
| FLI1 | Positive (nuclear) |
| Vimentin | Positive |
| NSE, S100, synaptophysin | Variably positive (neuroectodermal differentiation) |
| Keratin/EMA | Negative |
| Desmin, myogenin, MyoD1 | Negative |
| LCA (CD45) | Negative |
| TdT | Negative |
Molecular/Genetics
- >90% carry a balanced translocation: t(11;22)(q24;q12) generating EWS-FLI1 fusion gene (most common, ~85%)
- Other ETS partners: EWS-ERG [t(21;22)], EWS-ETV1, EWS-ETV4
- Aggressive variant: CIC-DUX4 fusion
- Other somatic mutations: TP53, CDKN2A, STAG2
- Cell of origin: likely mesenchymal stem cells or primitive neuroectodermal cells
Diagnosis confirmed by molecular techniques: FISH, RT-PCR, or NGS for EWSR1 rearrangement.
Differential Diagnosis
Ewing sarcoma belongs to the "Small Round Blue Cell Tumors" (SRBCT) of childhood. This is the critical differential grouping.
Primary SRBCT Differential
| Tumor | Key Distinguishing Features |
|---|---|
| Lymphoma of bone (Primary) | LCA (CD45)+, B/T cell markers+; no EWSR1 rearrangement; cells more dispersed, not cohesive |
| Neuroblastoma | Age <5 typically; adrenal/paraspinal; Homer-Wright rosettes more prominent + neuropil; NB84+, chromogranin+, synaptophysin+; MYCN amplification; urinary catecholamines elevated |
| Rhabdomyosarcoma (embryonal/alveolar) | Desmin+, myogenin+, MyoD1+; MYOD1 or PAX3/PAX7-FOXO1 fusions; rhabdoid cytoplasm/cross-striations |
| Small-cell osteosarcoma | Produces osteoid matrix (key distinction); no EWSR1 translocation; alkaline phosphatase elevated |
| Mesenchymal chondrosarcoma | Cartilaginous matrix islands; SOX9+; NKX2.2-; HEY1-NCOA2 fusion |
| Wilms tumor (nephroblastoma) | Renal origin; triphasic pattern (blastema, epithelial, stromal); WT1+; not a bone tumor |
| Medulloblastoma | CNS location; neuronal rosettes; synaptophysin+; various molecular subgroups |
| Retinoblastoma | Ocular location; flexner-wintersteiner rosettes; RB1 mutation |
Other Important Differentials (Clinical/Radiological)
| Condition | Key Points |
|---|---|
| Osteomyelitis | Clinically very similar (fever, leukocytosis, elevated ESR/CRP, local pain/swelling); culture positive; no EWSR1; average misdiagnosis delay ~34 weeks |
| Langerhans Cell Histiocytosis | Eosinophilic granuloma; bone disruption faster (7-10 days vs 4-6 weeks for Ewing); Birbeck granules; CD1a+, S100+, langerin+ |
| Osteosarcoma | Osteoid production; metaphyseal; older peak age; no EWSR1 |
| Lymphoma | LCA+; no glycogen; molecular markers |
| Metastatic neuroblastoma to bone | Young children; catecholamines; adrenal primary |
| Leukemia (bone involvement) | Systemic; CBC abnormalities; marrow infiltration |
| Primitive neuroectodermal tumor (PNET) | Now classified within Ewing family; same EWSR1-ETS fusion; more neural differentiation; same treatment |
Note on PNET: The WHO currently classifies PNET and Ewing sarcoma together as the Ewing sarcoma family of tumors (ESFT), unified by their shared EWSR1-ETS molecular signature and treated identically.
Practical Diagnostic Approach
- H&E: Sheets of SRBCT with clear glycogen-rich cytoplasm
- PAS stain: Positive, diastase-sensitive (confirms glycogen)
- IHC panel: CD99 (diffuse membrane+), FLI1+, exclude LCA, desmin, keratin, chromogranin
- Molecular: FISH for EWSR1 break-apart / RT-PCR for EWS-FLI1 fusion transcript - gold standard for confirmation
Sources:
- Robbins & Kumar Basic Pathology, p. 786
- Rheumatology 2-Volume Set (Elsevier, 2022), p. 1982
- Campbell's Operative Orthopaedics 15th Ed 2026, pp. 1221-1243
- Robbins, Cotran & Kumar Pathologic Basis of Disease, p. (pediatric tumors chapter)
Rhabdomyosarcoma of lower leg bone histopathology description
Finding Sources
Searching PubMed
"rhabdomyosarcoma" histopathology extremity
Reading File
Reading File
Loading Image
Reading File
Reading File
Reading File
Excellent - I now have comprehensive, multi-source data. Here is the complete answer:
Rhabdomyosarcoma of the Lower Leg - Histopathology
Key Context: Which Subtype Affects the Lower Leg?
RMS of the extremities (including the lower leg) is predominantly the alveolar subtype in children/adolescents, while pleomorphic RMS is the characteristic adult extremity type. The embryonal subtype predominantly affects head/neck and genitourinary sites. All subtypes can secondarily involve bone from an adjacent soft tissue primary.
Gross Pathology
- Alveolar RMS: Firm, less gelatinous/myxoid mass; usually within or adjacent to muscle; may invade periosteum and cortex secondarily
- Pleomorphic RMS: Large, infiltrative mass, often deep in muscle compartments of the extremity
- Both types show hemorrhage and necrosis
Microscopic Histopathology (by Subtype)
1. Alveolar RMS (Most common in extremities, including lower leg)
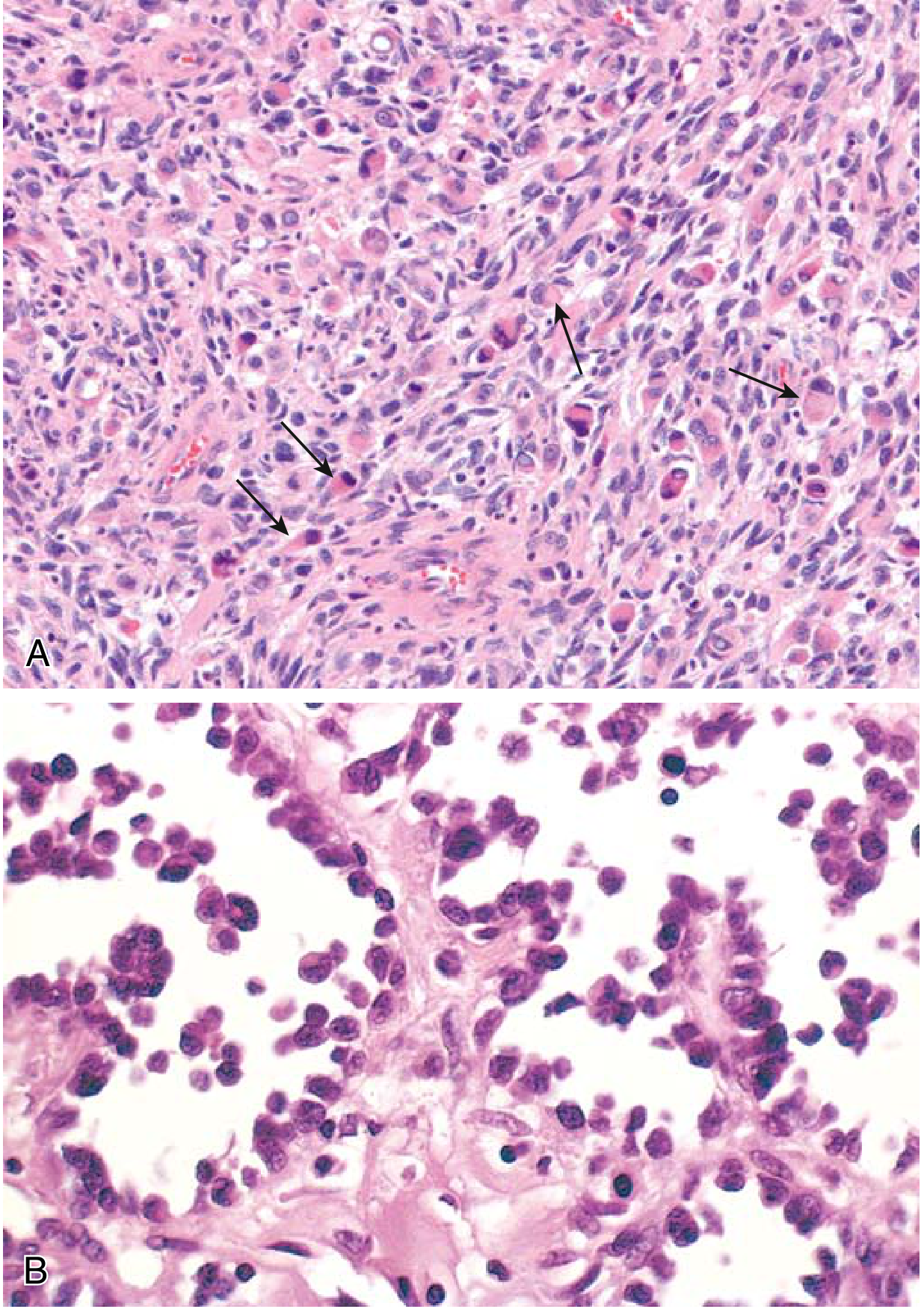
FIG. 19.44 A & B (Robbins Basic Pathology) - (A) Embryonal subtype with rhabdomyoblasts (arrows); (B) Alveolar subtype with dyscohesive round cells lining fibrovascular septa:
| Feature | Description |
|---|---|
| Architecture | Alveolar pattern - fibrous septa divide cells into nests/clusters resembling pulmonary alveoli |
| Predominant cell | Round cells with scanty eosinophilic cytoplasm; only minimally cohesive |
| Cell cohesion | Central loss of cohesion within nests; peripheral cells adhere to fibrovascular septa |
| Rhabdomyoblasts | Occasional - large round/elongated "strap"-shaped cells with deeply eosinophilic granular cytoplasm; cross-striations may be visible (reflecting sarcomere formation) |
| Nuclei | Round, hyperchromatic; prominent nucleoli |
| Multinucleated giant cells | Present but usually inconspicuous |
| Mitoses | High mitotic index |
| Necrosis | Frequent; in rare cases viable cells are virtually absent |
| Stroma | Fibrovascular septa dividing cell clusters |
2. Pleomorphic RMS (Classic adult extremity type)
| Feature | Description |
|---|---|
| Architecture | Spindle-shaped cells in parallel and interlacing bundles |
| Cells | Large, sometimes multinucleate, bizarre eosinophilic tumor cells |
| Characteristic cells | Strap-shaped and racquet-shaped rhabdomyoblasts - most easily identifiable here |
| Giant cells | Prominent multinucleated giant cells |
| Mitoses | Prominent mitotic activity |
| Cross-striations | May be seen in strap cells (confirms skeletal muscle lineage) |
3. Embryonal RMS (Less common in lower leg; included for completeness)
| Feature | Description |
|---|---|
| Architecture | Alternating cellular and myxoid zones; parallel bundles |
| Cells | Small, round or spindle-shaped with hyperchromatic nuclei; abundant cytoplasm |
| Stroma | Myxoid background; more gelatinous grossly |
| Rhabdomyoblasts | Present with straplike cytoplasm and visible cross-striations |
The Rhabdomyoblast - Diagnostic Hallmark
The rhabdomyoblast is the essential diagnostic cell in all RMS subtypes:
- Large round, spindle, or elongated "strap"-shaped cell
- Deeply eosinophilic granular cytoplasm rich in thick and thin filaments
- Cross-striations may be visible (sarcomere formation - pathognomonic)
- Nuclei: eccentric, with prominent nucleoli
- Cells may also show ovoid, pleomorphic, or clear cytomorphology
Immunohistochemistry (IHC)
| Marker | Result | Notes |
|---|---|---|
| Myogenin (MYF4) | Strongly positive | Highest sensitivity + specificity for RMS; nuclear staining |
| MyoD1 | Strongly positive | Nuclear; excellent specificity for skeletal muscle lineage |
| Desmin | Positive | Less specific (also positive in leiomyosarcoma) |
| Muscle-specific actin (MSA) | Positive | |
| Vimentin | Positive | Non-specific |
| CD99 | Sometimes positive in ERMS and ARMS | Can cause confusion with Ewing sarcoma |
| LCA (CD45) | Negative | Excludes lymphoma |
| S100, HMB-45 | Negative | Excludes melanoma |
| Keratin | Negative | Excludes carcinoma |
Key point: Myogenin and MyoD1 are the highest diagnostic value markers for distinguishing RMS from other SRBCTs in the lower leg.
Molecular Genetics
| Subtype | Translocation | Fusion Gene |
|---|---|---|
| Alveolar RMS | t(2;13)(q35;q14) | PAX3-FOXO1 (~60%) |
| Alveolar RMS | t(1;13)(p36;q14) | PAX7-FOXO1 (~20%) |
| Embryonal RMS | 11p15 LOH | Heterogeneous; no specific fusion |
| Pleomorphic RMS | Complex karyotype | TP53, RB1 alterations |
- Approximately 80% of ARMS carry PAX3-FOXO1 or PAX7-FOXO1 fusion
- Molecular confirmation by FISH or RT-PCR is used alongside IHC
Special Note on "Lower Leg Bone" Involvement
True primary intraosseous RMS is extremely rare. When RMS involves lower leg bone, it is almost always by:
- Direct extension from an adjacent soft tissue mass into periosteum and cortex
- Metastatic disease to bone marrow (particularly in ARMS)
Metastases in RMS occur to lungs, lymph nodes, and bone marrow. The ARMS with PAX3-FOXO1 fusion has a particularly aggressive course with higher metastatic risk.
Sources:
- Robbins & Kumar Basic Pathology, p. 802 (FIG. 19.44)
- Fitzpatrick's Dermatology 2-Volume Set, pp. 2173-2174
- Campbell's Operative Orthopaedics 15th Ed 2026, pp. 1278-1279
Malignant ewings sarcoma histopathology description
This question was covered in detail in our earlier session. Here is the complete histopathology description from the authoritative textbooks already consulted:
Ewing Sarcoma - Histopathology Description
Gross Pathology
- Location: Usually diaphyseal, arising in the medullary cavity; most commonly femur, pelvis, tibia, fibula
- Texture: Soft, tan-white tumor
- Cut surface: Frequent areas of hemorrhage and necrosis
- Extent: Invades cortex, periosteum, and surrounding soft tissue
- Periosteal reaction: Advancing tumor stimulates layers of reactive bone - the classic "onion-skin" appearance on imaging; a "sunburst" pattern may also occur
Microscopic Histopathology
FIG. 19.23 (Robbins & Kumar Basic Pathology) - Sheets of small round cells with minimal clear cytoplasm:
60x photomicrograph (Rheumatology, Elsevier) - Small round blue cell tumor with fibrovascular spaces:
Feature-by-Feature Description
| Histological Feature | Description |
|---|---|
| Overall pattern | Broad sheets/diffuse arrangement of uniform tumor cells |
| Cell size | Small round cells - slightly larger and more cohesive than lymphocytes |
| Cytoplasm | Scant; characteristically appears clear/pale due to abundant glycogen |
| Glycogen | PAS-positive, diastase-sensitive - one of the most reliable histochemical features |
| Nuclei | Round, uniform; fine "salt-and-pepper" chromatin |
| Nucleoli | Usually inconspicuous |
| Cell cohesion | Cells are cohesive (unlike lymphoma) |
| Homer-Wright rosettes | May be present - circular groupings of cells around a central fibrillary core; indicates neuroectodermal differentiation |
| Matrix | No osteoid, bone, or cartilage production (critical negative feature) |
| Mitoses | Variable |
| Necrosis | Common, often extensive |
| Stroma | Scant fibrovascular stroma; delicate vessels between cell sheets |
Summary: The "3 Unities" of Ewing Sarcoma Histology
- Uniform cells - monotonous population without pleomorphism
- Round nuclei - no angulation or irregular contours
- Clear cytoplasm - glycogen-rich, PAS+
Immunohistochemistry
| Marker | Result |
|---|---|
| CD99 (MIC2) | Strongly positive - diffuse membranous staining (most characteristic) |
| FLI1 | Positive (nuclear) |
| NKX2.2 | Positive - highly specific for Ewing |
| Vimentin | Positive |
| NSE, synaptophysin, S100 | Variably positive (neuroectodermal component) |
| Desmin, Myogenin, MyoD1 | Negative (excludes RMS) |
| LCA (CD45) | Negative (excludes lymphoma) |
| Keratin/EMA | Negative |
| TdT | Negative |
Molecular Confirmation (Gold Standard)
- >90%: Balanced translocation t(11;22)(q24;q12) → EWS-FLI1 fusion (~85%)
- Other variants: EWS-ERG [t(21;22)], EWS-ETV1, EWS-ETV4
- Confirmed by: FISH (EWSR1 break-apart probe), RT-PCR, or NGS
- The EWSR1-ETS fusion protein dysregulates transcription leading to uncontrolled growth
Pathogenesis
- Cell of origin: likely mesenchymal stem cells or primitive neuroectodermal cells
- The chimeric EWS/FLI1 protein binds chromatin and dysregulates transcription through uncertain mechanisms, driving abnormal differentiation and uncontrolled proliferation
Sources:
- Robbins & Kumar Basic Pathology, p. 786 (FIG. 19.23)
- Rheumatology 2-Volume Set (Elsevier 2022), p. 1982 (FIG. E218.38)
- Campbell's Operative Orthopaedics 15th Ed 2026, pp. 1221-1222
This is a shared conversation. Sign in to Orris to start your own chat.