Neurohormonal reflex of heart
Neurohormonal Reflexes of the Heart
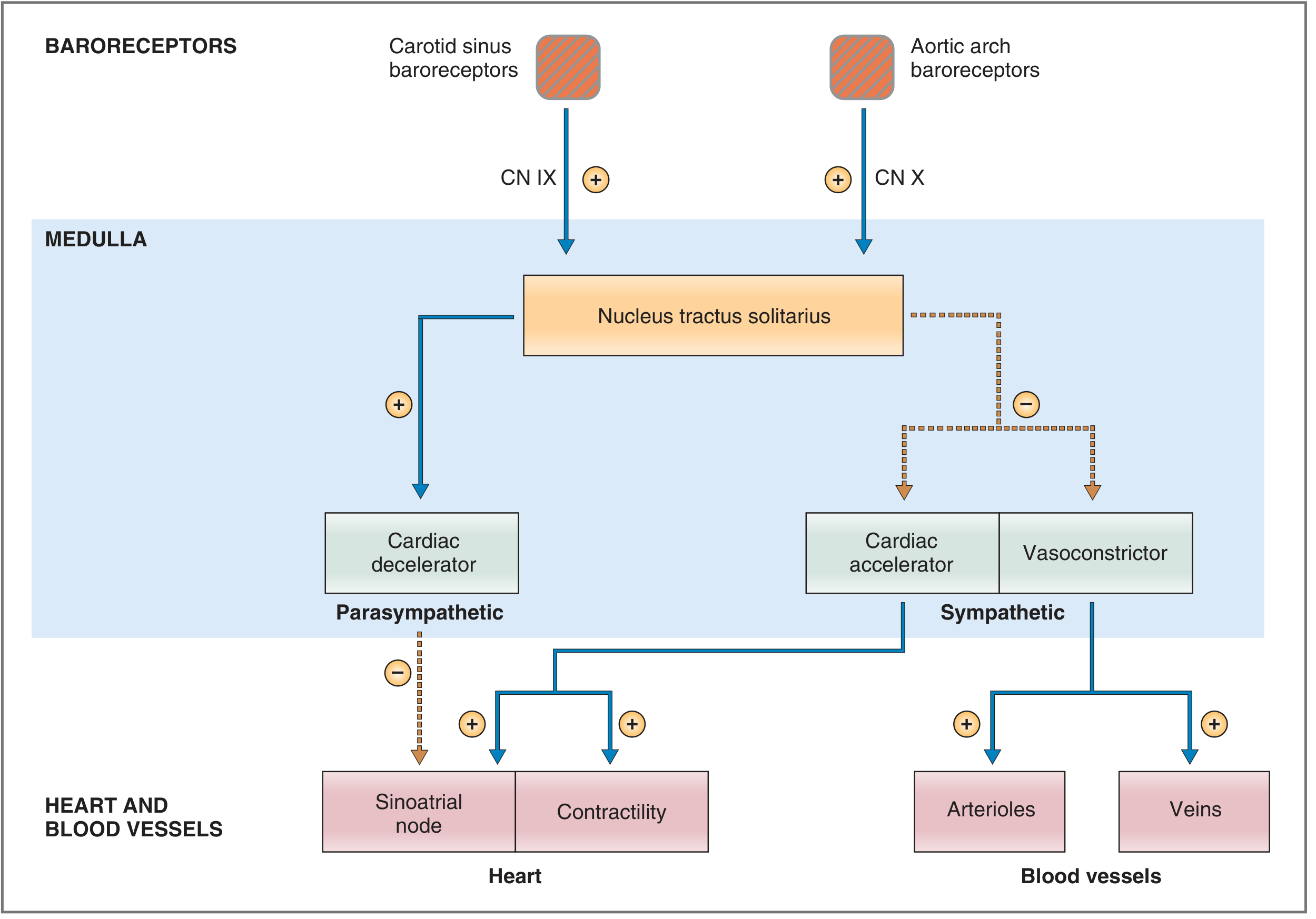
1. The Baroreceptor (Pressoreceptor) Reflex
Sensors - Baroreceptors
- Carotid sinus - at the bifurcation of the common carotid artery (responds to both increases and decreases in pressure)
- Aortic arch - responds primarily to increases in arterial pressure (operates ~30 mmHg higher than carotid receptors)
Afferent Pathway
| Receptor Location | Nerve | Destination |
|---|---|---|
| Carotid sinus | Hering's nerve → Glossopharyngeal (CN IX) | Nucleus tractus solitarius (NTS), medulla |
| Aortic arch | Vagus nerve (CN X) | NTS, medulla |
Brain Stem Cardiovascular Centers
- Vasoconstrictor center (C1) - upper medulla/lower pons; sympathetic efferents cause arteriolar and venous constriction
- Cardiac accelerator center - sympathetic efferents increase SA node firing rate, AV conduction velocity, and myocardial contractility
- Cardiac decelerator center - parasympathetic (vagal) efferents slow the SA node
Integrated Response
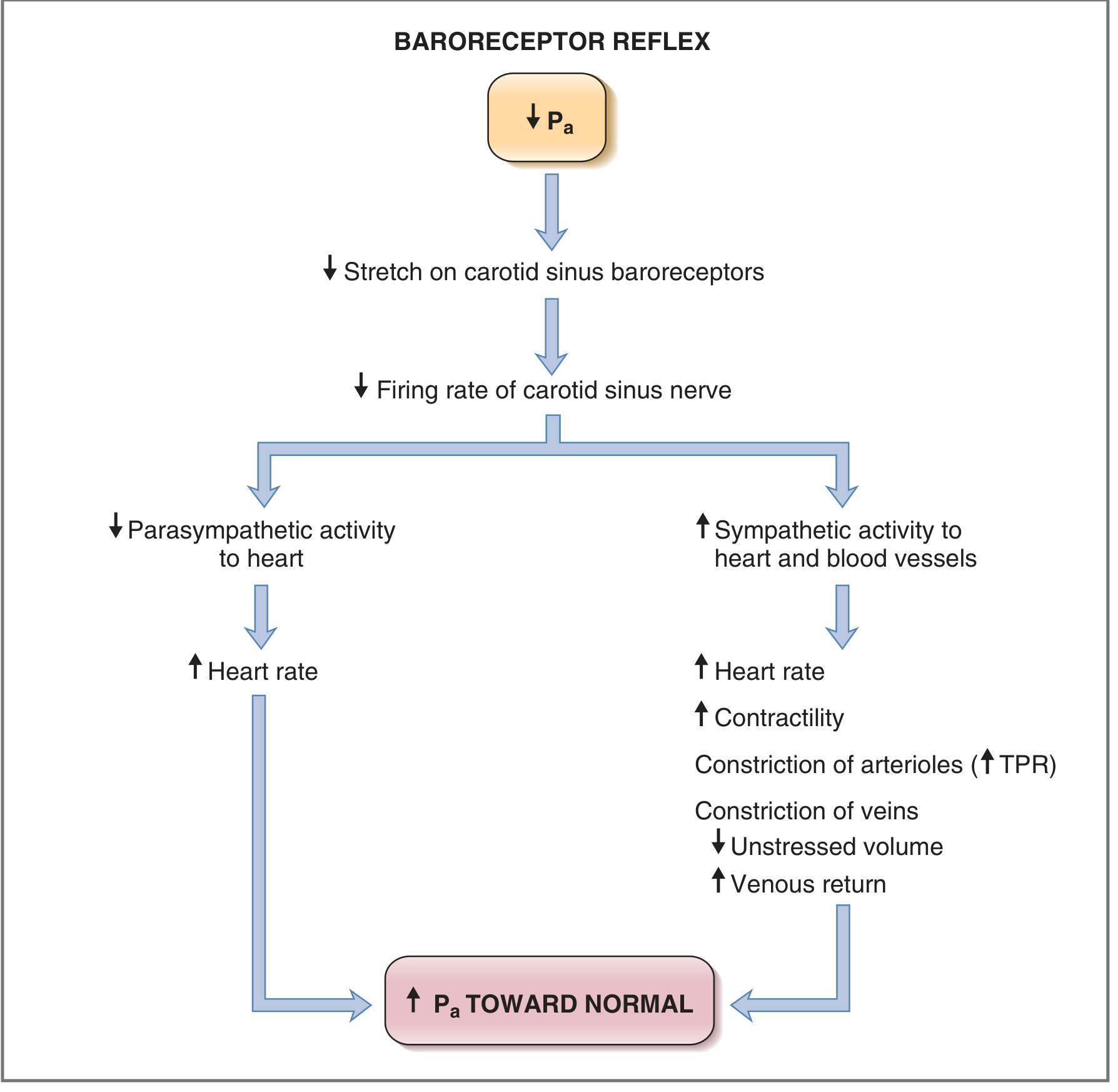
- Increased stretch → increased firing of baroreceptor afferents
- NTS increases parasympathetic outflow + decreases sympathetic outflow
- Heart rate decreases (vagal slowing of SA node)
- Cardiac contractility decreases (reduced stroke volume)
- Arteriolar vasodilation (decreased TPR)
- Venodilation (increased unstressed volume, decreased venous return)
- Net effect: Cardiac output falls, TPR falls → Pa returns to normal
- Decreased stretch → decreased baroreceptor firing
- NTS decreases parasympathetic outflow + increases sympathetic outflow
- Heart rate increases, contractility increases → cardiac output increases
- Arteriolar vasoconstriction (increased TPR)
- Venoconstriction (decreased unstressed volume → increased stressed volume and venous return via Frank-Starling mechanism)
- Net effect: Pa restored toward normal
Clinical Applications
- Orthostatic hypotension: On standing, pressure in the head and upper body falls. The immediate baroreceptor reflex triggers strong sympathetic discharge to prevent loss of consciousness.
- Valsalva maneuver: Forced expiration against closed glottis → increased intrathoracic pressure → decreased venous return → decreased cardiac output → decreased Pa → baroreceptor reflex triggers tachycardia and vasoconstriction. On release, venous return surges → Pa rises → reflex bradycardia.
- Chronic hypertension: Baroreceptors reset to the higher pressure; reflex now defends the elevated set point rather than correcting it. This leads to perioperative circulatory instability.
- Anesthetic effects: Volatile anesthetics (especially halothane) inhibit the heart rate component. ACE inhibitors, calcium channel blockers, and phosphodiesterase inhibitors lessen the pressor response.
2. Chemoreceptor Reflex
- Sensors: Chemosensitive cells in the carotid bodies and aortic body, responding to:
- PaO₂ < 50 mmHg (hypoxia)
- Acidosis (low pH)
- Afferent pathway: Hering's nerve (branch of CN IX) and CN X to the chemosensitive area of the medulla
- Response: Primarily increases respiratory drive; also stimulates cardiovascular centers to increase heart rate and contractility
- Persistent hypoxia: The CNS itself is directly stimulated, causing a global increase in sympathetic activity
3. Bainbridge Reflex (Atrial Stretch Reflex)
- Sensors: Stretch receptors in the right atrial wall and the cavoatrial junction
- Stimulus: Increased right-sided filling pressure (volume loading)
- Afferent pathway: Vagal afferents to the medullary cardiovascular center
- Response: Inhibition of parasympathetic activity → increased heart rate (tachycardia)
- There is also a direct mechanical effect: atrial stretch accelerates the SA node directly
- The magnitude of the heart rate change depends on the baseline heart rate before stimulation
- Function: Prevents venous congestion by speeding heart rate when the right heart is overfilled
4. Bezold-Jarisch Reflex (Cardio-Inhibitory Reflex)
- Sensors: Chemoreceptors and mechanoreceptors within the left ventricular wall
- Stimulus: Noxious ventricular stimuli (ischemia, infarction, thrombolysis, revascularization, or overdistension)
- Afferent pathway: Unmyelinated vagal C-fibers → increases parasympathetic tone
- Response triad: Hypotension + Bradycardia + Coronary artery vasodilation
- Considered a cardioprotective reflex by reducing myocardial oxygen demand during ischemia
- Modulated by natriuretic peptide receptors (ANP/BNP) - less pronounced in cardiac hypertrophy or atrial fibrillation
- Clinically implicated in vasovagal syncope and post-MI bradycardia
5. Cushing Reflex
- Stimulus: Raised intracranial pressure → cerebral ischemia at the medullary vasomotor center
- Initial response: Sympathetic activation → increased heart rate, arterial pressure, and contractility (to restore cerebral perfusion)
- Secondary response: The resulting high vascular tone is sensed by baroreceptors → reflex bradycardia (Cushing's triad: hypertension + bradycardia + abnormal respirations)
6. Oculocardiac Reflex
- Stimulus: Pressure on the globe of the eye or traction on extraocular muscles
- Afferent pathway: Short/long ciliary nerves → ophthalmic division of trigeminal nerve (CN V₁) → Gasserian ganglion → medulla
- Response: Increased parasympathetic tone → bradycardia (and occasionally asystole)
- Clinically important in ophthalmic surgery
Summary Table
| Reflex | Sensor Location | Stimulus | Afferent Nerve | Response |
|---|---|---|---|---|
| Baroreceptor | Carotid sinus, aortic arch | Stretch (↑ or ↓ BP) | CN IX, CN X | Negative feedback restores BP |
| Chemoreceptor | Carotid/aortic bodies | Hypoxia, acidosis | CN IX, CN X | ↑ HR, contractility, ventilation |
| Bainbridge | Right atrial wall / cavoatrial junction | ↑ Venous return/filling pressure | Vagal afferents | ↑ HR (tachycardia) |
| Bezold-Jarisch | Left ventricular wall | Ischemia, toxins, overdistension | Vagal C-fibers | Bradycardia + hypotension + coronary dilation |
| Cushing | Medullary vasomotor center | Cerebral ischemia (raised ICP) | CNS direct | Hypertension → reflex bradycardia |
| Oculocardiac | Extraocular muscles, globe | Ocular pressure/traction | CN V₁ → medulla | Bradycardia |
- Costanzo Physiology, 7th Edition - Baroreceptor Reflex chapter
- Guyton and Hall Textbook of Medical Physiology - Baroreceptor Arterial Pressure Control chapter
- Miller's Anesthesia, 10th Edition - Baroreceptor, Chemoreceptor, Bainbridge, Bezold-Jarisch, and other cardiac reflexes
Why subendocardial position most susceptible to ischemia
Why the Subendocardium Is Most Susceptible to Ischemia
1. Last to Receive Blood from Epicardial Coronary Vessels
"Irreversible injury of ischemic myocytes first occurs in the subendocardial zone. This region is especially susceptible to ischemia because it is the last area to receive blood delivered by the epicardial vessels."
- Robbins & Cotran Pathologic Basis of Disease
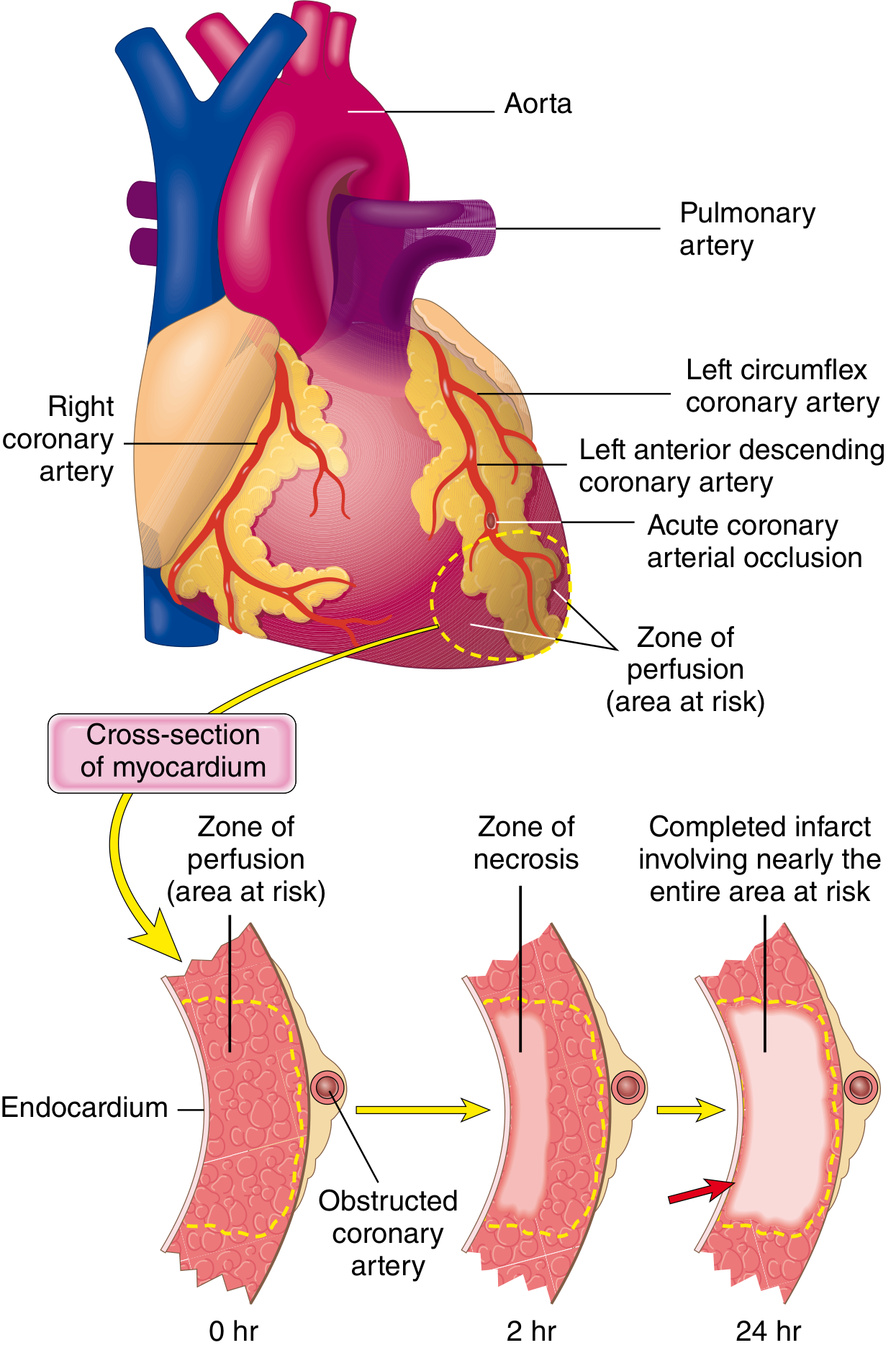
2. Coronary Perfusion of the LV is Almost Entirely Diastolic - and the Subendocardium Bears the Brunt
- Subendocardial vessels are nearly completely occluded during systole
- The epicardial vessels continue to receive some flow throughout the cardiac cycle
- 70-80% of LV coronary blood flow occurs during diastole
- Adequate diastolic perfusion pressure
- Adequate duration of diastole
- Intact autoregulatory reserve
3. Coronary Perfusion Pressure Is Lowest at the Subendocardium
Coronary Perfusion Pressure = Aortic Diastolic Pressure - LV End-Diastolic Pressure (LVEDP)
"Elevations in LVEDP impede subendocardial blood flow."
- Miller's Anesthesia, 10th Edition
4. Highest Wall Stress - Laplace's Law
- The subendocardium faces the full intracavitary pressure directly
- The inner-layer fibers bear more load than the outer-layer fibers in a curved pressure vessel
- Greater myocardial oxygen demand (myofilaments require more ATP to develop tension)
- Greater extravascular compressive force on intramural vessels (impeding inflow)
"Wall stress is not uniformly distributed across LV thickness in the intact heart; it is greatest in the subendocardium and progressively declines toward the epicardial surface. The subendocardium is exposed to marked increases in LV pressure and myocardial oxygen demand that render it more susceptible to ischemia."
- Barash Clinical Anesthesia, 9th Edition
5. Effect of Tachycardia
- Shortens diastole disproportionately (reducing the only window in which subendocardial perfusion occurs)
- Increases myocardial oxygen demand globally
Summary: The Perfect Storm
| Factor | Effect on Subendocardium |
|---|---|
| Epicardial vessel anatomy | Last to receive blood (terminal distribution) |
| Systolic compression | Vessels nearly occluded during systole (only diastolic window for flow) |
| LVEDP elevation | Directly reduces subendocardial perfusion pressure |
| Wall stress (Laplace) | Highest in subendocardium → highest O₂ demand + highest compressive force |
| Tachycardia | Shortens diastole, exhausts subendocardial flow reserve |
| Limited collaterals | Terminal arterioles, poor collateral network |
- Robbins & Cotran Pathologic Basis of Disease - Progression of Myocardial Necrosis
- Barash Clinical Anesthesia, 9th Edition - Coronary Physiology and Laplace's Law
- Miller's Anesthesia, 10th Edition - Determinants of Myocardial Oxygen Supply and Demand
Streptokinase in mi
Streptokinase in Myocardial Infarction
Origin and Classification
Mechanism of Action
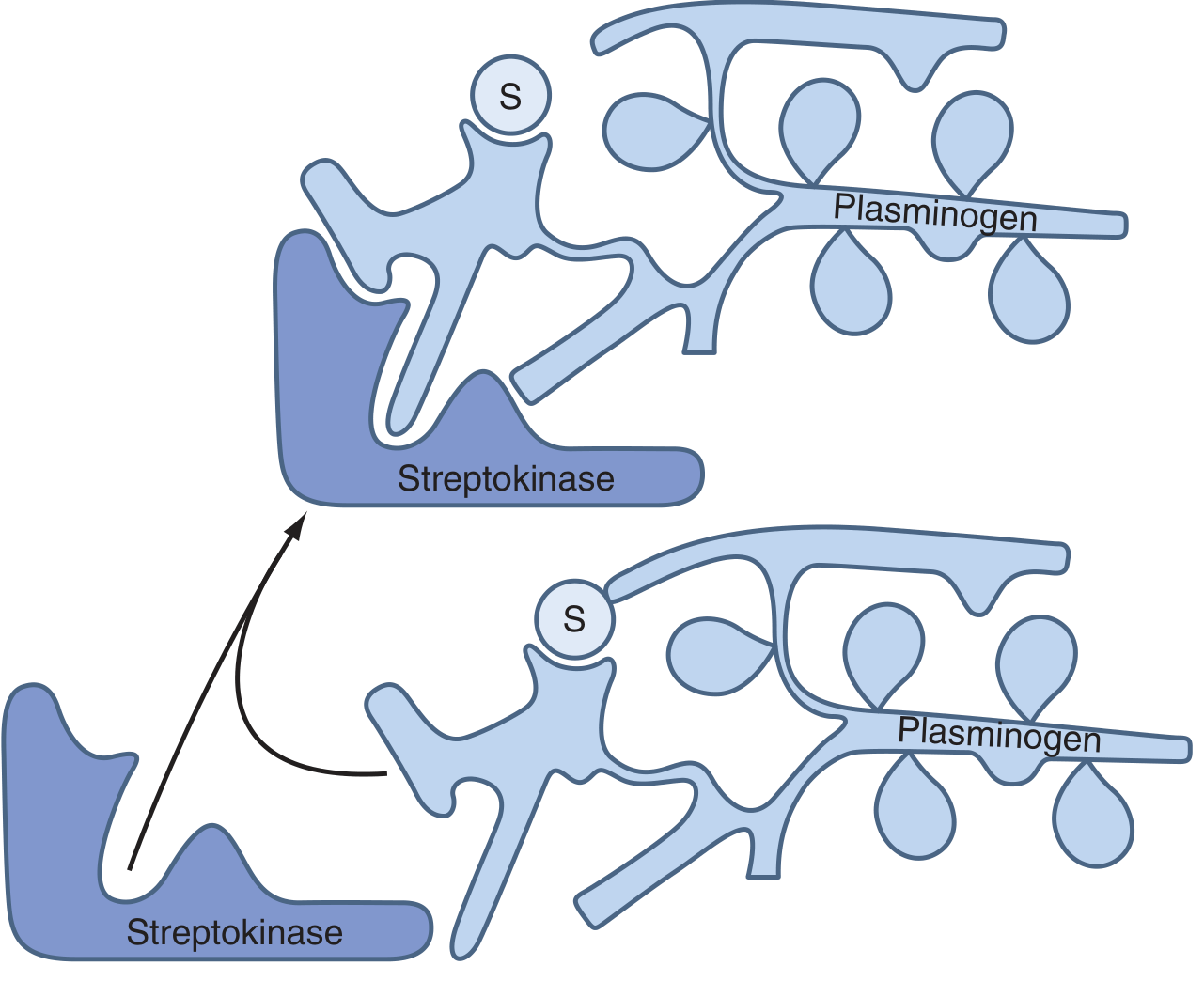
Key mechanistic distinction - No Fibrin Specificity
- Circulating fibrinogen is also degraded (not just clot fibrin)
- Fibrin degradation products (FDPs) accumulate
- Bleeding risk is generalized, not just at the clot site
Pharmacokinetics
| Parameter | Value |
|---|---|
| Serum half-life | ~23 minutes |
| Duration of fibrinolytic effect | Up to 24 hours |
| Route | IV infusion |
| Standard dose in STEMI | 1.5 million units IV over 30-60 minutes |
Use in STEMI
- STEMI (ST elevation ≥1 mm in ≥2 contiguous leads, or new LBBB)
- Symptom onset within 12 hours
- No access to primary PCI within 90-120 minutes of first medical contact
- No absolute contraindications
Adverse Effects
1. Bleeding
2. Allergic Reactions (~5-6% of patients)
- Rash, fever, chills, rigors
- Anaphylaxis (rare but life-threatening) Patients with prior streptococcal infection may already have pre-formed antibodies that reduce effectiveness or trigger severe reactions.
3. Transient Hypotension (common)
- Leg elevation
- IV fluids
- Low-dose vasopressors (dopamine or norepinephrine) if needed
Antibody Formation and Re-administration
- Antibodies to streptokinase begin forming approximately 5 days after treatment and persist for up to 6 months
- These antibodies can neutralize subsequent doses and reduce effectiveness
- Re-treatment with streptokinase is contraindicated within 6 months of prior administration
- Streptokinase should also not be given within 12 months of a streptococcal infection (due to high pre-existing antibody titers)
- If re-thrombolysis is needed, a fibrin-specific agent (alteplase, tenecteplase) must be used instead
Comparison with Other Thrombolytics
| Property | Streptokinase (1st gen) | Alteplase/tPA (2nd gen) | Tenecteplase (3rd gen) |
|---|---|---|---|
| Origin | Streptococci | Recombinant human | Recombinant (modified tPA) |
| Mechanism | Indirect (complex with plasminogen) | Direct cleavage of plasminogen | Direct (modified tPA) |
| Fibrin specificity | None (systemic lysis) | Moderate | High |
| Antigenicity | Yes (allergic reactions in ~5%) | No (<2%) | No |
| Re-administration | Contraindicated (6 months) | Allowed | Allowed |
| Dose | 1.5 MU over 30-60 min | Weight-based infusion over 60-90 min | Single weight-based bolus |
| Heparin after | Delayed | Given promptly after | Given promptly after |
| Cost | Low | Higher | Higher |
Absolute Contraindications to Fibrinolysis (including Streptokinase)
- Any prior intracranial hemorrhage
- Ischemic stroke within 3 months
- Known intracranial neoplasm or AV malformation
- Active internal bleeding (excluding menses)
- Suspected aortic dissection
- Significant closed-head trauma within 3 months
- Intracranial or intraspinal surgery within 2 months
- Severe uncontrolled hypertension (>180/110 mmHg unresponsive to treatment)
Clinical Evidence
- GISSI-1 trial - demonstrated 18% relative reduction in 35-day mortality with streptokinase vs placebo in STEMI
- ISIS-2 trial - showed combined aspirin + streptokinase reduced mortality by ~42% compared to placebo; each drug alone reduced mortality by ~23-25%
- Braunwald's Heart Disease, 2nd Vol - Streptokinase mechanism and clinical use
- Harrison's Principles of Internal Medicine 22E (2025) - Streptokinase pharmacology
- Tintinalli's Emergency Medicine - First-generation thrombolytics in acute coronary syndromes
"A 65 years old man who is a known case of diabetes mellitus presents with dizziness as soon as he wakes from bed i n the morning. On examination blood pressure recorded in the supine position was 140/90 mmHg and on standing immediately was 110/70 mmHg. (1+4+5=10) (a) Name the clinical condition patient is suffering from? Describe briefly the short-term regulation o f blood pressure? 10) Explain the determinants o f mean arterial pressure"
Why cardiac muscle can not be tetanized
Why Cardiac Muscle Cannot Be Tetanized
What is Tetanus?
- The skeletal muscle action potential is very short (~1-2 ms)
- The mechanical twitch lasts much longer (~100 ms)
- So a second stimulus can arrive and fire a new action potential while the muscle is still contracting
The Core Reason: Prolonged Action Potential = Prolonged Absolute Refractory Period
Cardiac Action Potential Phases
| Phase | Name | Ion Movement | Duration |
|---|---|---|---|
| 0 | Upstroke | Fast Na⁺ influx | ~1 ms |
| 1 | Early rapid repolarization | K⁺ efflux (transient outward) | Brief |
| 2 | Plateau | Slow Ca²⁺ influx via L-type channels | 200-300 ms |
| 3 | Final repolarization | K⁺ efflux (delayed rectifiers) | ~100 ms |
| 4 | Resting potential | Na⁺-K⁺ ATPase restores balance | Stable at -80 to -90 mV |
"In contrast to axonal action potentials, cardiac action potentials include a plateau phase that lasts 0.2 to 0.3 s. Whereas the action potential for skeletal muscle and nerves is due exclusively to the opening of voltage-gated sodium channels, in cardiac muscle, the action potential is initiated by voltage-gated sodium channels (the spike) and maintained primarily by voltage-gated calcium channels (the plateau)."
- Morgan & Mikhail's Clinical Anesthesiology, 7th Edition
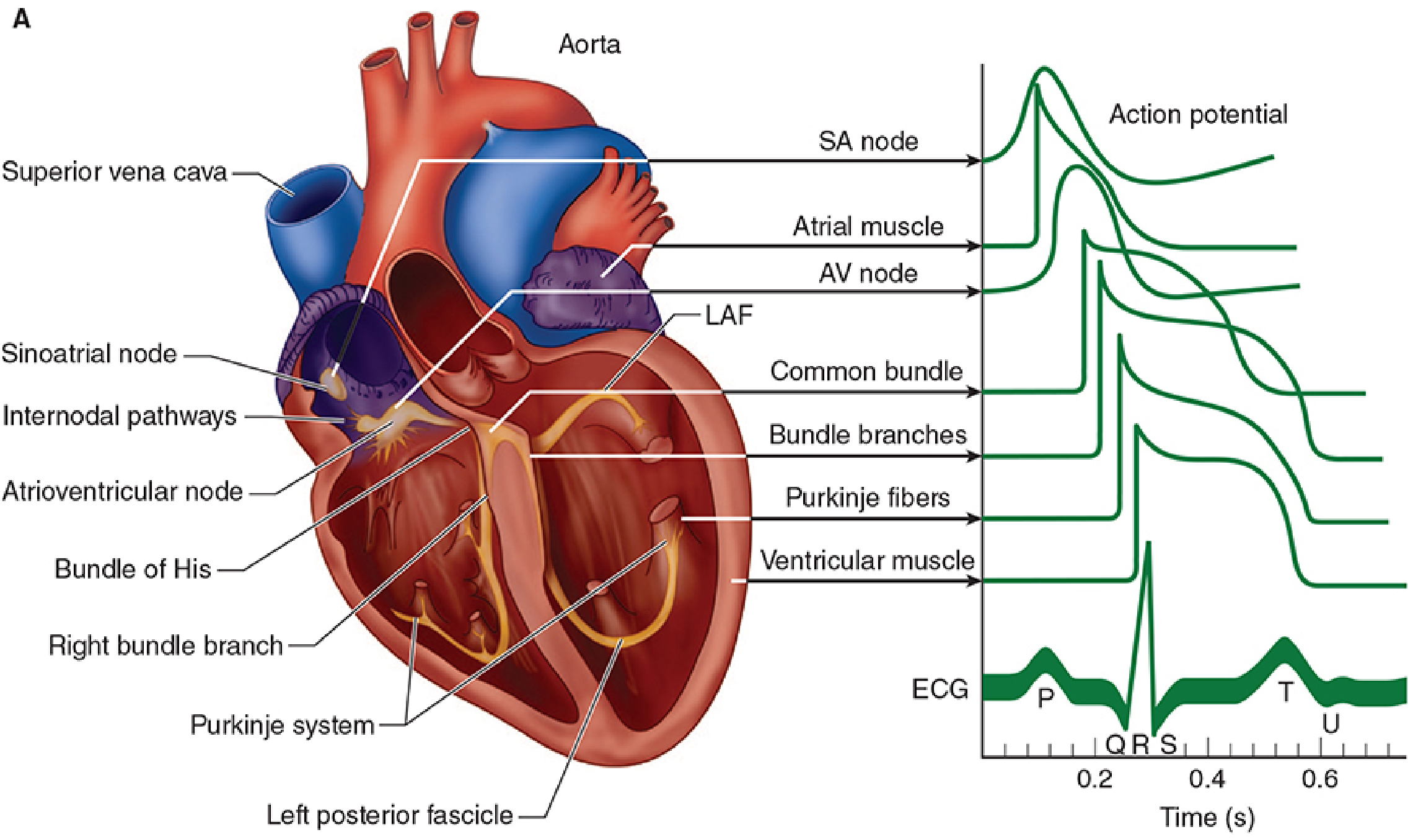
Why the Plateau Prevents Tetanus
- Voltage-gated Na⁺ channels are in their inactivated (closed) state
- They cannot reopen until the membrane repolarizes sufficiently (back toward -60 mV)
- The plateau (Phase 2) keeps the membrane depolarized for 200-300 ms, holding Na⁺ channels in an inactivated state the entire time
- The ARP therefore lasts almost as long as the entire mechanical contraction (~250-300 ms in ventricular muscle)
| Parameter | Skeletal Muscle | Cardiac Muscle |
|---|---|---|
| Action potential duration | ~1-2 ms | ~250-300 ms |
| Mechanical twitch duration | ~100 ms | ~300 ms |
| Absolute refractory period | ~1-2 ms | ~250 ms |
| Can a 2nd stimulus fire during twitch? | Yes - tetanus possible | No - ARP covers the contraction |
"Because it has a prolonged action potential, cardiac muscle cannot contract in response to a second stimulus until near the end of the initial contraction. Therefore, cardiac muscle cannot be tetanized like skeletal muscle."
- Ganong's Review of Medical Physiology, 26th Edition
The Ionic Basis: Why Is the Cardiac Action Potential So Long?
- After the initial Na⁺ spike (Phase 0), the membrane potential would rapidly repolarize as in a nerve - BUT
- L-type Ca²⁺ channels then open slowly and remain open for a prolonged period
- The inward Ca²⁺ current (depolarizing) balances the outward K⁺ current, producing the plateau
- The Ca²⁺ influx also triggers calcium-induced calcium release (CICR) from the sarcoplasmic reticulum, which is what actually causes muscle contraction
- Only when L-type Ca²⁺ channels inactivate (and delayed rectifier K⁺ channels activate) does Phase 3 repolarization begin
"Sustained depolarization during the plateau (Phase 2) is due primarily to the activity of calcium channels... cardiac calcium channels activate and inactivate in a manner similar to sodium channels, but in the case of the L-type channel, the transitions occur more slowly and at more positive potentials."
- Katzung's Basic and Clinical Pharmacology, 16th Edition
Functional and Teleological Significance
-
Pumping function requires complete relaxation: The heart fills with blood during diastole. If the ventricle could be tetanized, it would stay contracted, never relax, never fill, and cardiac output would fall to zero.
-
Protection from arrhythmia: The long refractory period prevents re-entrant excitation during the vulnerable period of the cycle.
-
Rate limiting by the AV node: The AV node's refractory period limits conduction to ~230 impulses/min in adults. Ventricular rates exceeding this are only seen in pathological states like ventricular tachycardia.
-
Relative refractory period (RRP): There is a brief period at the end of Phase 3 when the membrane is partially repolarized and a stronger-than-normal stimulus can fire a new action potential. A stimulus during this "vulnerable period" (corresponding to the peak of the T wave on ECG) can trigger dangerous ventricular fibrillation - the clinical basis of the "R-on-T phenomenon".
Summary Diagram (Conceptual)
Cardiac:
|←——— AP (250-300 ms) ———→|
|←————— ARP (~250 ms) ————→|
|←——————— Contraction (300 ms) ——————→|
A 2nd stimulus arriving during the ARP fires NOTHING.
Muscle CANNOT summate → Cannot tetanize.
Skeletal:
|← AP (1-2 ms)→|
|←ARP(~2ms)→|
|←———— Contraction (100 ms) ————————→|
A 2nd stimulus arrives HERE ↑ (during contraction, well after ARP).
Muscle summation occurs → Tetanus IS possible.
- Ganong's Review of Medical Physiology, 26th Edition - Length of Systole & Diastole
- Morgan & Mikhail's Clinical Anesthesiology, 7th Edition - Cardiac Action Potentials
- Katzung's Basic and Clinical Pharmacology, 16th Edition - Cardiac action potential ionic basis
"A motor cyclist met with a road traffic accident and was admitted in the causality. Clinical examination revealed, cold and clammy extremities apid thready pulse, and a blood pressure of 70/50mm of Hg. Based on the above, answer the following (1+1+2+4+2=10) I. What is the probable clinical condition? Il. Define shock. III. Name the different stages of shock. IV. Describe the compensatory mechanisms in shock. V. What i s neurogenic shock?"
Structured Answer: Shock in a Road Traffic Accident (10 marks)
Part I (1 mark): Probable Clinical Condition
- Cold, clammy extremities - intense peripheral vasoconstriction shunting blood to vital organs
- Rapid thready pulse - compensatory tachycardia with reduced stroke volume
- BP 70/50 mmHg - severe hypotension indicating decompensated shock
Part II (1 mark): Definition of Shock
- It is defined by inadequate tissue perfusion, not simply by hypotension
- The result is cellular hypoxia → anaerobic glycolysis → lactic acid production → lactic acidosis
- Without treatment, it progresses to irreversible organ damage and death
Part III (2 marks): Stages of Shock
Stage 1: Compensated Shock (Pre-shock)
- Blood volume/cardiac output is reduced, but the body's compensatory mechanisms maintain blood pressure and vital organ perfusion
- No overt organ dysfunction at this stage
- Subtle findings: mild tachycardia, mild anxiety, cool peripheries
- Labs may show mild elevation of creatinine, troponin, or lactate
- Fully reversible with prompt treatment
Stage 2: Decompensated Shock (True / Progressive Shock)
- Compensatory mechanisms are overwhelmed
- Blood pressure falls, tissue hypoperfusion becomes clinically manifest
- Frank organ dysfunction develops: oliguria (kidneys), confusion (brain), metabolic acidosis
- Lactic acidosis worsens
- Still potentially reversible with aggressive resuscitation
Stage 3: Irreversible Shock (Refractory Shock)
- Prolonged severe ischemia causes permanent cellular and organ damage
- Cell death in vital organs (heart, kidney, brain, liver, gut)
- Gut barrier failure → bacterial translocation → sepsis
- Multisystem organ dysfunction syndrome (MODS)
- Not reversible even with aggressive treatment
- Death results from multiorgan failure
"Regardless of type, shock progresses through a continuum of three stages: compensated shock (preshock), shock (decompensated shock), and irreversible shock. If untreated, the patient will progress to the third phase of irreversible shock. At this point, the organ dysfunction is permanent and often the patient progresses to multisystem organ dysfunction."
- Harrison's Principles of Internal Medicine, 22nd Edition
Part IV (4 marks): Compensatory Mechanisms in Shock
1. Neural Compensatory Mechanisms (Immediate - seconds)
- Fall in arterial pressure → decreased stretch on carotid sinus and aortic arch baroreceptors → decreased firing in afferent nerves (CN IX, X) → decreased inhibition of medullary vasomotor center
- Result: Massive sympathetic outflow + decreased parasympathetic tone
| Target | Effect | Purpose |
|---|---|---|
| SA node | ↑ Heart rate (tachycardia) | ↑ Cardiac output |
| Myocardium | ↑ Contractility | ↑ Stroke volume |
| Arterioles | Vasoconstriction (skin, gut, muscle) | ↑ TPR, redirect blood to heart/brain |
| Veins | Venoconstriction | ↓ Unstressed volume, ↑ venous return |
| Adrenal medulla | ↑ Epinephrine + norepinephrine release | Amplifies all the above |
2. Chemoreceptor Activation (Immediate-early)
- As blood pressure falls below ~50 mmHg, carotid and aortic body chemoreceptors sense local hypoxia → further amplify sympathetic vasoconstrictor output
- With severe brain ischemia, direct stimulation of the CNS vasomotor center causes an even more powerful sympathetic surge (Cushing response)
3. Hormonal Compensatory Mechanisms (Minutes to hours)
- Reduced renal perfusion + sympathetic stimulation of juxtaglomerular cells → ↑ Renin secretion
- Renin → Angiotensin I → ACE → Angiotensin II
- Angiotensin II effects:
- Potent arteriolar vasoconstriction (↑ TPR)
- Stimulates aldosterone secretion → Na⁺ and water retention by kidney → ↑ blood volume
- Stimulates ADH release
- Released from posterior pituitary in response to reduced atrial stretch (detected by low-pressure atrial receptors)
- Causes:
- Renal water reabsorption (↓ urine output) → ↑ blood volume
- Vasoconstriction at high concentrations
- Released from adrenal medulla via sympathetic activation
- Amplify tachycardia, vasoconstriction, and cardiac contractility
4. Transcapillary Fluid Shift (Minutes to hours)
- Arteriolar constriction → ↓ capillary hydrostatic pressure
- Interstitial fluid is drawn into the capillaries by the net oncotic pressure gradient (Starling forces)
- This hemodilution (auto-transfusion) can restore up to 75% of shed blood volume within one hour
- This is why hemorrhaged soldiers arrive with diluted (low hematocrit) blood
↓ Blood Volume / ↓ BP
↓
Baroreceptor + Chemoreceptor activation
↓
Sympathetic activation
├── ↑ HR + ↑ Contractility → ↑ CO
├── Arteriolar vasoconstriction → ↑ TPR
├── Venoconstriction → ↑ Venous return
└── Adrenal medulla → Epinephrine/NE
↓
RAAS → Angiotensin II + Aldosterone → Na/H₂O retention
ADH → Water retention
Transcapillary refill → ↑ Plasma volume
↓
Attempt to restore MAP = CO × TPR
Part V (2 marks): Neurogenic Shock
Mechanism
- Normally, the sympathetic nervous system maintains arteriolar and venous tone
- In spinal cord injury (cervical or high thoracic), sympathetic outflow below the lesion is completely lost
- This causes:
- Massive arteriolar vasodilation → ↓ SVR → ↓ blood pressure
- Venous pooling (↑ venous capacitance) → ↓ venous return → ↓ cardiac output
- Loss of sympathetic input to the heart → no compensatory tachycardia
- Loss of input to the adrenal medulla → no catecholamine surge
Causes
- Spinal cord trauma (vertebral fracture at C-spine or high T-spine)
- Spinal cord neoplasm
- Spinal or epidural anesthesia extending too high
- Severe brain injury / brainstem injury (with vasomotor paralysis)
- Deep general anesthesia (depresses vasomotor center)
Classic Clinical Features (Distinguishes it from other shock types)
| Feature | Neurogenic Shock | Hypovolemic Shock |
|---|---|---|
| BP | ↓ | ↓ |
| Heart rate | Bradycardia | Tachycardia |
| Skin | Warm and dry (vasodilated) | Cold and clammy |
| SVR | ↓↓ | ↑↑ |
| Cardiac output | ↓ | ↓ |
Treatment
- Volume resuscitation first (restores venous return)
- Vasopressors (e.g., norepinephrine, phenylephrine) if hypotension persists - to restore vascular tone
- In pure neurogenic shock without hypovolemia, vasopressors may also be used to improve heart rate and contractility (norepinephrine or dopamine)
- Maintain spinal cord perfusion pressure to prevent secondary ischemic injury
"The classic description of neurogenic shock consists of decreased blood pressure associated with bradycardia (absence of reflexive tachycardia due to disrupted sympathetic discharge), warm extremities (loss of peripheral vasoconstriction), motor and sensory deficits indicative of spinal cord injury."
- Schwartz's Principles of Surgery, 11th Edition
- Harrison's Principles of Internal Medicine, 22nd Edition (2025) - Stages of Shock
- Schwartz's Principles of Surgery, 11th Edition - Neurogenic Shock
- Guyton and Hall Textbook of Medical Physiology - Neurogenic causes of distributive shock
- Medical Physiology (Boron & Boulpaep) - Cardiovascular reflexes in hemorrhage
Write a short note on mean electrical axis of heart.
Short Note: Mean Electrical Axis of the Heart
Definition
"The preponderant direction of the potential during depolarization from the base to the apex of the heart is called the mean electrical axis of the ventricles."
- Guyton and Hall Textbook of Medical Physiology
Anatomical Basis
- Septal depolarization (Phase 1): Depolarization begins in the interventricular septum, spreading from left to right and anteriorly (small vector)
- Ventricular free wall depolarization (Phase 2): Simultaneous activation of both ventricles, but the left ventricle is electrically dominant (greater muscle mass), so the net vector points leftward, downward, and posteriorly
Normal Values
- The anatomical position of the heart (tilted to the left in the chest)
- The electrical dominance of the left ventricle
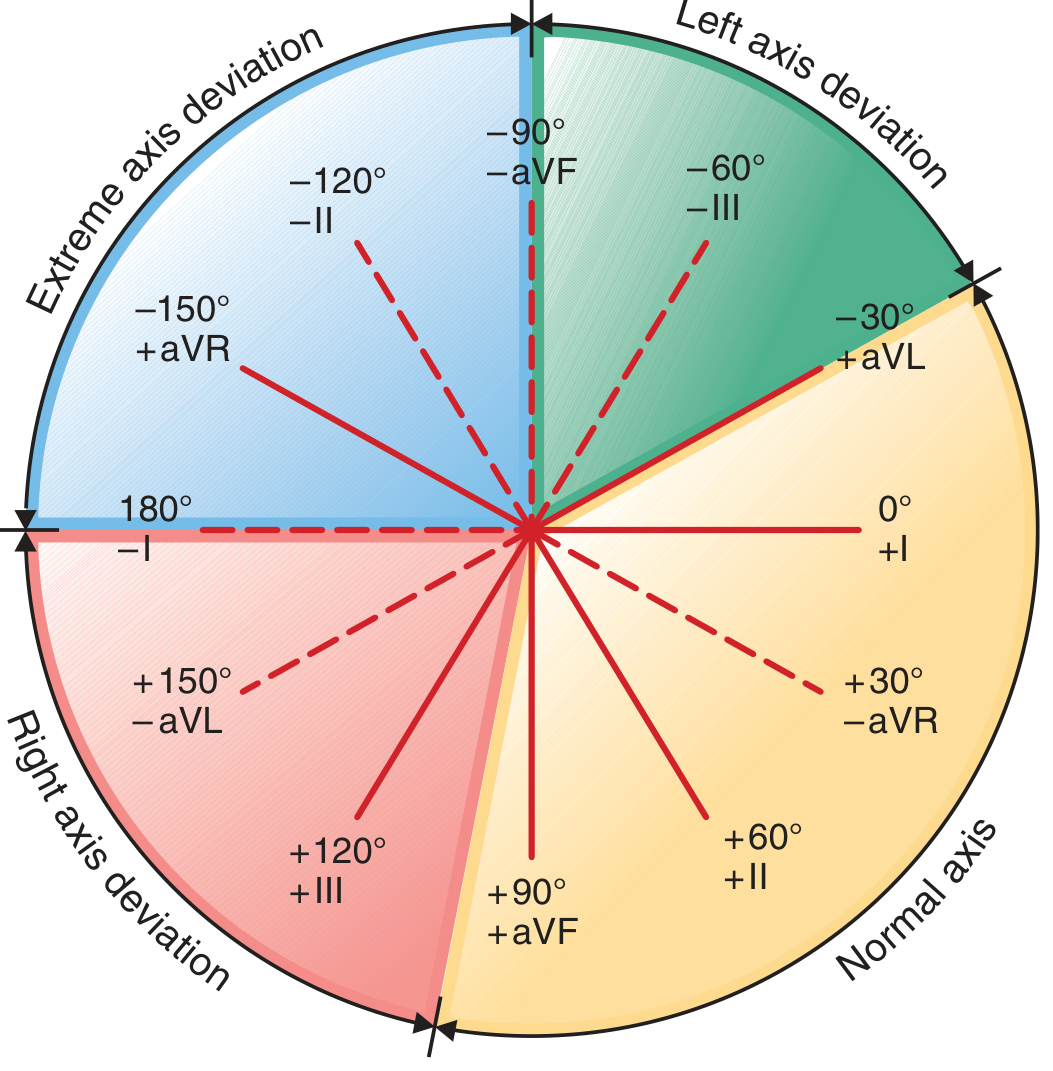
The Hexaxial Reference System
| Lead | Angle |
|---|---|
| Lead I | 0° |
| Lead II | +60° |
| Lead III | +120° |
| aVR | -150° |
| aVL | -30° |
| aVF | +90° |
How to Determine the Mean Electrical Axis
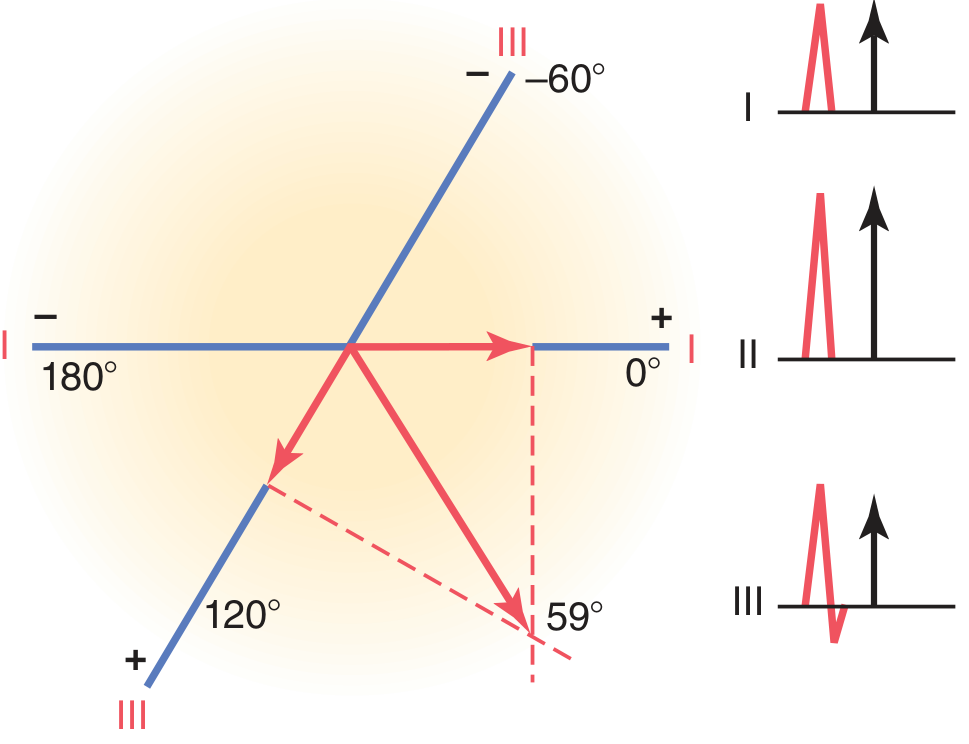
Method 1: Two-Lead (Perpendicular) Method (Guyton)
- Measure the net QRS deflection (positive area minus negative area) in Lead I and Lead III
- Plot each net value on its respective lead axis on the hexaxial diagram
- Draw perpendiculars from the tip of each plotted vector
- The point where the perpendiculars intersect = the tip of the MEA vector
- Draw the MEA vector from the origin to this intersection point - the angle it makes = MEA
Method 2: Isoelectric Lead Method (Quick Clinical Shortcut)
- Find the lead where the QRS is most isoelectric (biphasic / net zero)
- The MEA lies at 90° to that lead's axis (i.e., the MEA is perpendicular to the isoelectric lead)
- Determine the direction (positive or negative 90°) by checking which direction gives a positive deflection in any other lead
Classification of Axis
| Axis Range | Classification |
|---|---|
| -30° to +90° | Normal axis |
| -30° to -90° | Left axis deviation (LAD) |
| +90° to +180° | Right axis deviation (RAD) |
| -90° to ±180° | Extreme axis deviation ("northwest axis") |
Causes of Axis Deviation
Left Axis Deviation (axis more negative than -30°)
- Left ventricular hypertrophy (hypertension, aortic stenosis, aortic regurgitation) - increased LV muscle mass generates dominant leftward vectors
- Left anterior fascicular block (LAFB) - most common cause of marked LAD (-45° to -90°)
- Left bundle branch block
- Inferior MI (loss of inferiorly directed vectors)
- Physiological: obese individuals, pregnancy, deep expiration (heart pushed upward/leftward)
Right Axis Deviation (axis more positive than +90°)
- Right ventricular hypertrophy (pulmonary stenosis, tetralogy of Fallot, pulmonary hypertension) - RV mass increases, pulling axis rightward
- Right bundle branch block
- Left posterior fascicular block (LPFB)
- Lateral MI (loss of laterally directed vectors)
- Dextrocardia
- Physiological: tall thin individuals, deep inspiration (heart hangs down and rightward)
Bundle Branch Block and Axis Deviation
- LBBB: Axis shifts leftward
- RBBB: Axis shifts rightward (axis may move toward +120° or further right)
Clinical Significance
| Condition | Axis Finding |
|---|---|
| Hypertension / Aortic stenosis | LAD (LVH) |
| Pulmonary hypertension / PE | RAD (RVH) |
| LAFB | Marked LAD (-45° to -90°) |
| Inferior MI | LAD (loss of inferior vectors) |
| Lateral MI | RAD (loss of lateral vectors) |
| Dextrocardia | Extreme right axis; lead I negative throughout |
| Pregnancy / Obesity | Physiologic LAD |
| WPW syndrome | Axis depends on accessory pathway location |
- Guyton and Hall Textbook of Medical Physiology - Mean Electrical Axis of the Ventricles
- Harrison's Principles of Internal Medicine, 22nd Edition - ECG Leads and QRS Complex
- Ganong's Review of Medical Physiology, 26th Edition - Bipolar limb leads and the cardiac vector