Teach me the basis of one medicine, pick one subject that you think you should choose for one Biotechnology student and
Reading File
Teach me the basis of one medicine, pick one subject that you think you would choose for one Biotechnology student and explain it to me in detail while including medical significance
Finding Sources
Searching the Web
pharmacology enzyme inhibition drug mechanisms biotechnology students
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Now I have everything I need. Here is your full, detailed lesson:
Pharmacology for Biotechnology Students: Enzyme Inhibition as a Drug Mechanism
Why this subject? As a biotech student, you already speak the language of proteins, active sites, binding kinetics, and molecular interactions. Enzyme inhibition sits right at the crossroads of biochemistry (which you know) and pharmacology (which you are learning). More than 30% of all approved drugs work by inhibiting a specific enzyme. Understanding this one concept unlocks the logic behind entire drug classes - from antibiotics to cancer therapies to blood pressure medications.
1. The Big Picture: Why Target Enzymes?
Enzymes catalyze reactions that keep cells alive. If a pathological process depends on a specific enzyme - whether it is a bacterial enzyme you want to destroy, a human enzyme running out of control in disease, or a viral enzyme needed for replication - you can design a molecule to shut it down.
As Goodman & Gilman's explains, well-known enzyme targets include:
- Angiotensin-converting enzyme (ACE) - inhibited by captopril, enalapril, lisinopril to lower blood pressure
- Cyclooxygenase (COX) - inhibited by aspirin and NSAIDs to reduce inflammation and pain
- Dihydrofolate reductase (DHFR) - inhibited by methotrexate (cancer) and trimethoprim (bacteria)
Any substance that decreases the velocity of an enzyme-catalyzed reaction is called an inhibitor (Lippincott Biochemistry, 8th Ed.).
2. Reversible vs. Irreversible Inhibition
The first critical distinction in pharmacology is whether the inhibitor binds permanently or temporarily.
Irreversible Inhibitors
These bind through covalent bonds. Once the inhibitor attaches, the enzyme is permanently disabled. The only way for the body to recover enzyme activity is to synthesize new enzyme protein.
- Example - Aspirin: Aspirin irreversibly inhibits COX-1 and COX-2 (cyclooxygenase enzymes). It acetylates a serine residue inside the enzyme's active site. Because platelets have no nucleus, they cannot make new COX. This is why aspirin's anti-platelet effect lasts the entire 7-10 day lifetime of the platelet - clinically important for stroke and heart attack prevention.
- Example - Lead poisoning: Lead (Pb²⁺) forms covalent bonds with sulfhydryl (-SH) groups of cysteine residues. It irreversibly inhibits ferrochelatase, the enzyme that inserts iron into protoporphyrin to make heme. The result is a buildup of protoporphyrin and anemia.
Biotech relevance: When designing a drug to covalently label an enzyme, you need to know which amino acid residue you are targeting (serine, cysteine, lysine, etc.) - this is exactly the kind of protein engineering question you will encounter in drug discovery.
Reversible Inhibitors
These bind via non-covalent bonds (hydrogen bonds, hydrophobic interactions, ionic bonds). Diluting the complex releases the inhibitor and restores enzyme activity. There are three main types:
3. The Three Types of Reversible Inhibition
A. Competitive Inhibition
The inhibitor looks like the substrate. It competes for the same active site.
- At low [substrate], the inhibitor often wins.
- At very high [substrate], you can flood the enzyme and outcompete the inhibitor.
- Result: Vmax is unchanged, but apparent Km increases.
Kinetically, this means the drug appears to lower the enzyme's affinity for its substrate (higher Km = lower affinity).
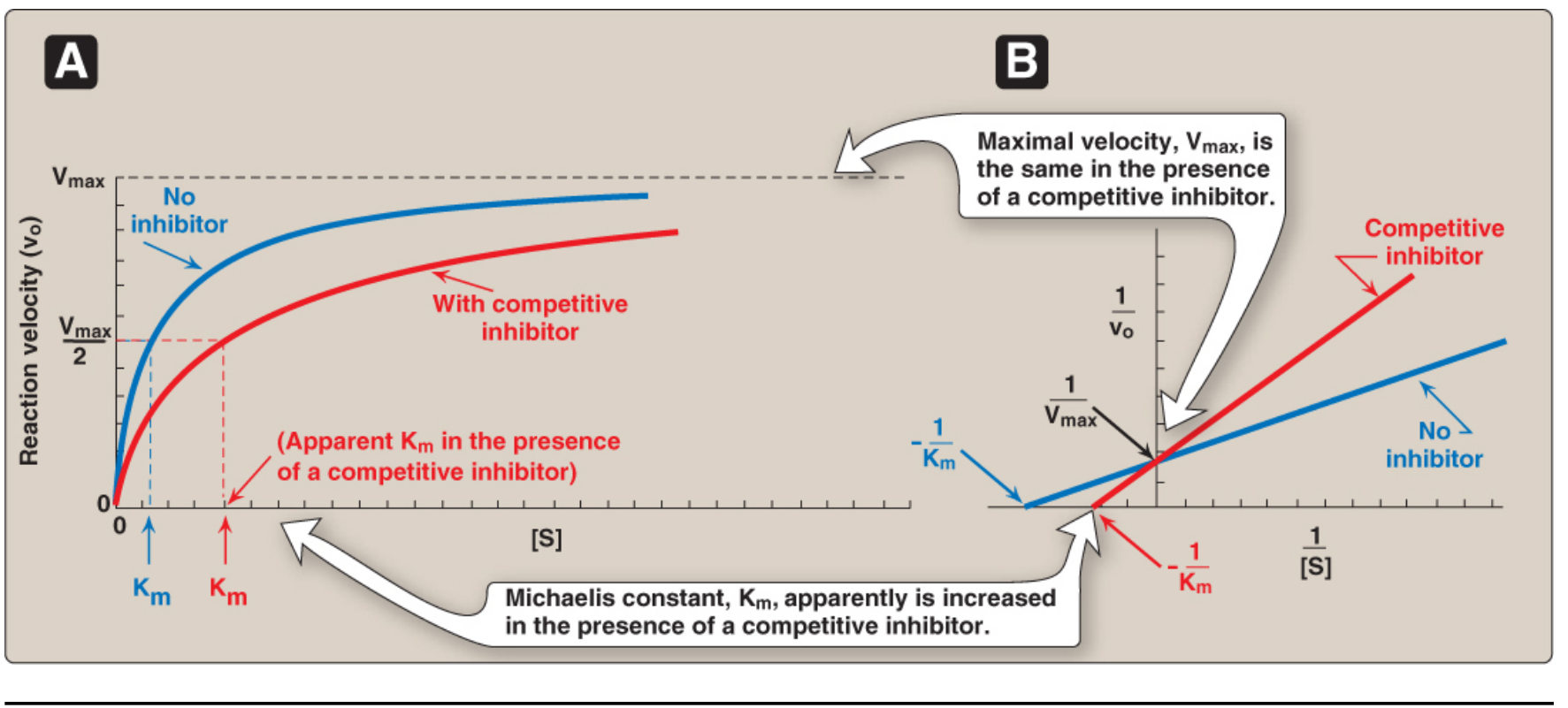
Clinical example - Statins: Statins (atorvastatin, rosuvastatin) competitively inhibit HMG-CoA reductase, the rate-limiting enzyme in cholesterol synthesis. They closely mimic the transition state of the natural substrate HMG-CoA. Because they bind with 1,000x higher affinity than the substrate itself, even small doses are highly effective. This is the basis of the most widely prescribed class of drugs in the world for lowering LDL cholesterol.
Clinical example - ACE inhibitors: Captopril competitively inhibits angiotensin-converting enzyme. This prevents conversion of angiotensin I → angiotensin II (a potent vasoconstrictor), causing blood vessel relaxation and lower blood pressure.
B. Noncompetitive Inhibition
The inhibitor binds at a different site from the active site (an allosteric site). It can bind to the free enzyme (E) or to the already-formed enzyme-substrate complex (ES). In either case, it reduces the enzyme's catalytic power without physically blocking substrate access.
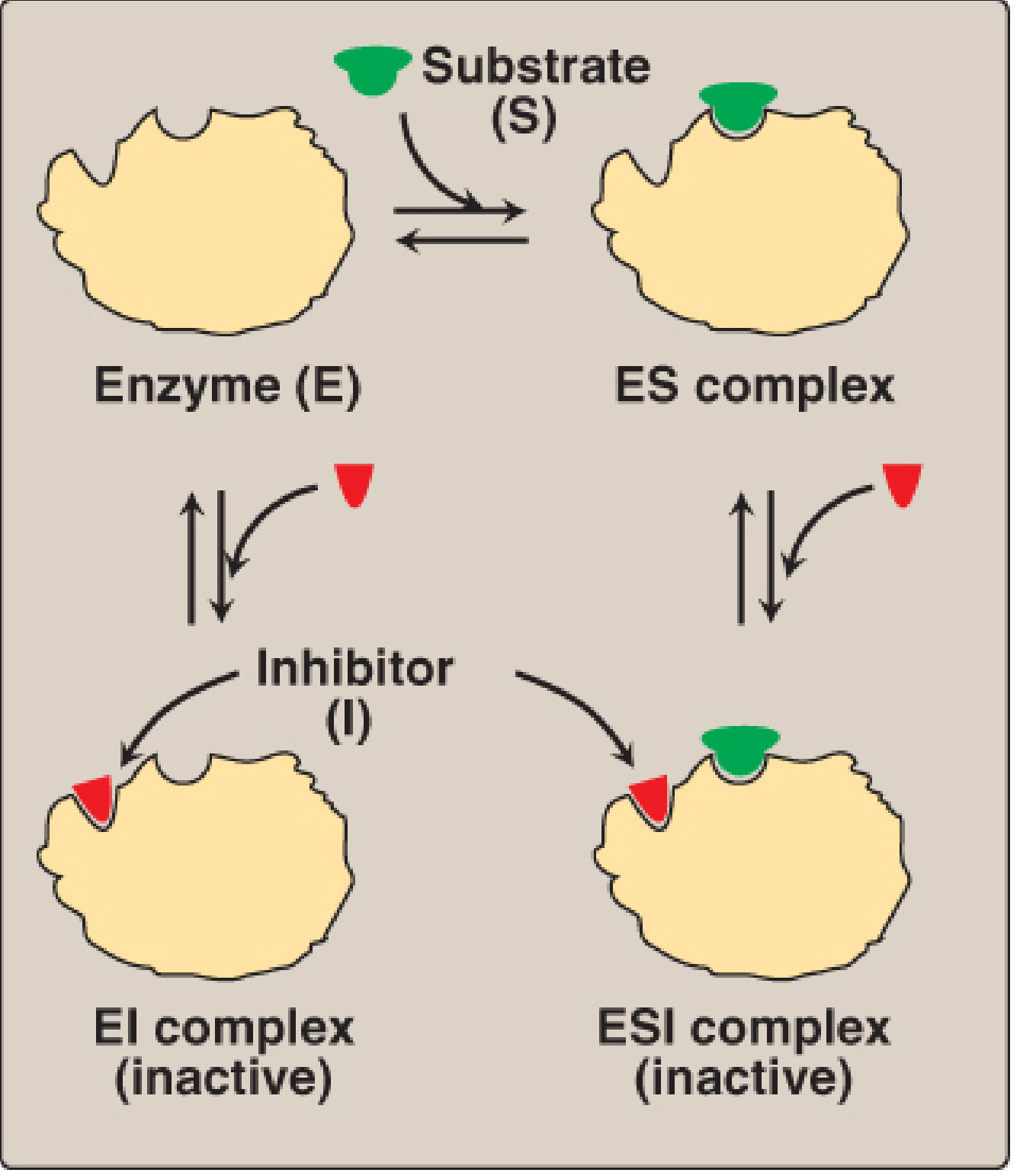
- The substrate can still bind (Km is unchanged).
- But maximum velocity drops: Vmax decreases, Km unchanged.
- You cannot overcome this by adding more substrate - flooding the active site does not help because the block is elsewhere.
Clinical/biotech relevance: Allosteric drugs are increasingly sought after in drug design because allosteric sites are often more unique to a specific enzyme isoform, allowing you to hit one target without affecting related enzymes. This is a major area of active research in the biotech industry.
C. Uncompetitive Inhibition
The inhibitor binds only to the ES complex (not to free enzyme). This is less common but clinically real.
- Both Vmax and Km decrease by the same factor.
- Paradoxically, adding more substrate makes inhibition worse (because more ES complex forms, giving the inhibitor more binding sites).
Example: Li⁺ (lithium) inhibits inositol monophosphatase uncompetitively. This is proposed as part of the mechanism by which lithium stabilizes mood in bipolar disorder.
4. Kinetics You Need to Know: Michaelis-Menten and Km
Since you have a biotech background, you are already familiar with Michaelis-Menten kinetics:
$$v_0 = \frac{V_{max} [S]}{K_m + [S]}$$
Key points:
- Km = substrate concentration at which the reaction is at half-Vmax. It reflects enzyme-substrate affinity (low Km = high affinity).
- Vmax = maximum velocity when all enzyme molecules are saturated with substrate.
- The Lineweaver-Burk (double-reciprocal) plot (1/v vs 1/[S]) turns the hyperbola into a straight line, making it easy to visually distinguish inhibitor types - this is the classic experimental tool used in biochemical pharmacology.
| Inhibitor Type | Vmax | Apparent Km |
|---|---|---|
| Competitive | Unchanged | Increased |
| Noncompetitive | Decreased | Unchanged |
| Uncompetitive | Decreased | Decreased |
5. Special Case: Suicide Inhibitors (Mechanism-Based Irreversible Inhibitors)
This is especially relevant in biotech drug design. A suicide inhibitor is a compound that looks normal - the enzyme actually begins to process it - but during catalysis it is converted into a reactive species that covalently destroys the enzyme from within.
Examples:
- Clavulanic acid: Bacteria produce beta-lactamase enzymes to destroy penicillin antibiotics. Clavulanic acid is a suicide inhibitor of beta-lactamase - it mimics penicillin, the enzyme tries to cleave it, and in doing so becomes permanently inactivated. Combined with amoxicillin (as Augmentin), it restores antibiotic effectiveness against resistant bacteria.
- Eflornithine: Inhibits ornithine decarboxylase (used to treat African sleeping sickness and excessive facial hair in women).
- 5-Fluorouracil (5-FU): A cancer drug that irreversibly inhibits thymidylate synthase, blocking DNA synthesis in tumor cells.
6. Drug-Receptor Pharmacodynamics: Beyond Enzymes
Enzyme inhibition is one category of drug mechanism. Drugs also act on:
- Receptors (G-protein coupled, ion channels, nuclear receptors) as agonists or antagonists
- Ion channels - voltage-gated Na⁺, K⁺, Ca²⁺ channels (local anesthetics, antiarrhythmics)
- Transporters - SSRIs (antidepressants) block serotonin reuptake transporters
All drug-receptor interactions share these key kinetic concepts with enzyme-inhibitor biochemistry - the same logic of Km, affinity, competition, and saturation applies. The concentration that produces 50% of maximal effect is called EC₅₀ (analogous to Km in enzymology).
7. Why This Matters for Biotechnology Specifically
| Biotechnology Application | Enzyme Inhibition Principle Used |
|---|---|
| Drug discovery / lead optimization | Designing competitive inhibitors mimicking transition states |
| Recombinant protein production | Using protease inhibitors to protect target proteins during purification |
| Biosensor design | Measuring inhibition of reporter enzymes to detect analytes |
| CRISPR & gene regulation | Inhibiting DNA repair enzymes to improve editing efficiency |
| Cancer immunotherapy | Checkpoint inhibitors block PD-L1/PD-1 (which act like enzyme-receptor inhibitors at the cell surface) |
| Antibiotic resistance research | Understanding beta-lactamase inhibition (clavulanate) to restore drug efficacy |
Summary
The subject of enzyme inhibition as a pharmacological mechanism gives you:
- A direct connection between your biochemistry training (enzymes, kinetics, protein structure) and clinical medicine
- A framework to understand dozens of drug classes immediately
- The intellectual foundation for drug design, where biotechnology and medicine collide
The three key drugs to remember as perfect examples of each inhibition type:
- Statins (competitive, HMG-CoA reductase) - cholesterol
- Aspirin (irreversible, COX) - pain/anti-platelet
- Augmentin/clavulanate (suicide inhibitor, beta-lactamase) - antibiotic resistance
Sources: Lippincott Illustrated Reviews: Biochemistry 8e, Goodman & Gilman's Pharmacological Basis of Therapeutics, Lippincott Illustrated Reviews: Pharmacology.
This is a shared conversation. Sign in to Orris to start your own chat.