Sickle cell anemia
Reading File
Finding Sources
Searching PubMed
"sickle cell disease"[MeSH Terms] AND treatment
Reading File
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Reading File
Reading File
Loading Image
Reading File
I now have an excellent body of information from multiple authoritative sources. Let me compile the full comprehensive answer.
Sickle Cell Anemia
Definition & Epidemiology
Sickle cell anemia (HbSS) is the most common familial hemolytic anemia and the prototypic hemoglobinopathy. It results from a single-point mutation in the β-globin gene. In the United States, approximately 8% of people of African descent are heterozygous carriers (sickle cell trait), and about 1 in 600 are homozygous (HbSS) and have the disease. The HbS allele reached high prevalence in equatorial Africa, parts of India, the Middle East, and southern Europe due to the selective advantage it confers against falciparum malaria in heterozygotes. - Robbins & Kumar Basic Pathology, p. 386
Genetics & Molecular Basis
- Inheritance: Autosomal recessive
- Mutation: A single nucleotide substitution (GAG → GTG) at codon 6 of β-globin, resulting in valine replacing glutamic acid at position 6
- This produces sickle hemoglobin (HbS) = α₂β₂^S
- Normal adult Hb composition: ~96% HbA (α₂β₂), ~3% HbA2 (α₂δ₂), ~1% HbF (α₂γ₂)
- In HbSS patients, HbA is completely replaced by HbS; heterozygotes retain ~60% HbA
Subtypes by severity:
| Genotype | Name | Severity |
|---|---|---|
| HbSS | Sickle cell anemia | Most severe |
| HbSβ⁰ thalassemia | Sickle-β⁰ thal | Most severe |
| HbSC | Sickle-hemoglobin C | Moderate |
| HbSβ⁺ thalassemia | Sickle-β⁺ thal | Milder |
- Harriet Lane Handbook, 23rd ed., p. 479
Pathogenesis
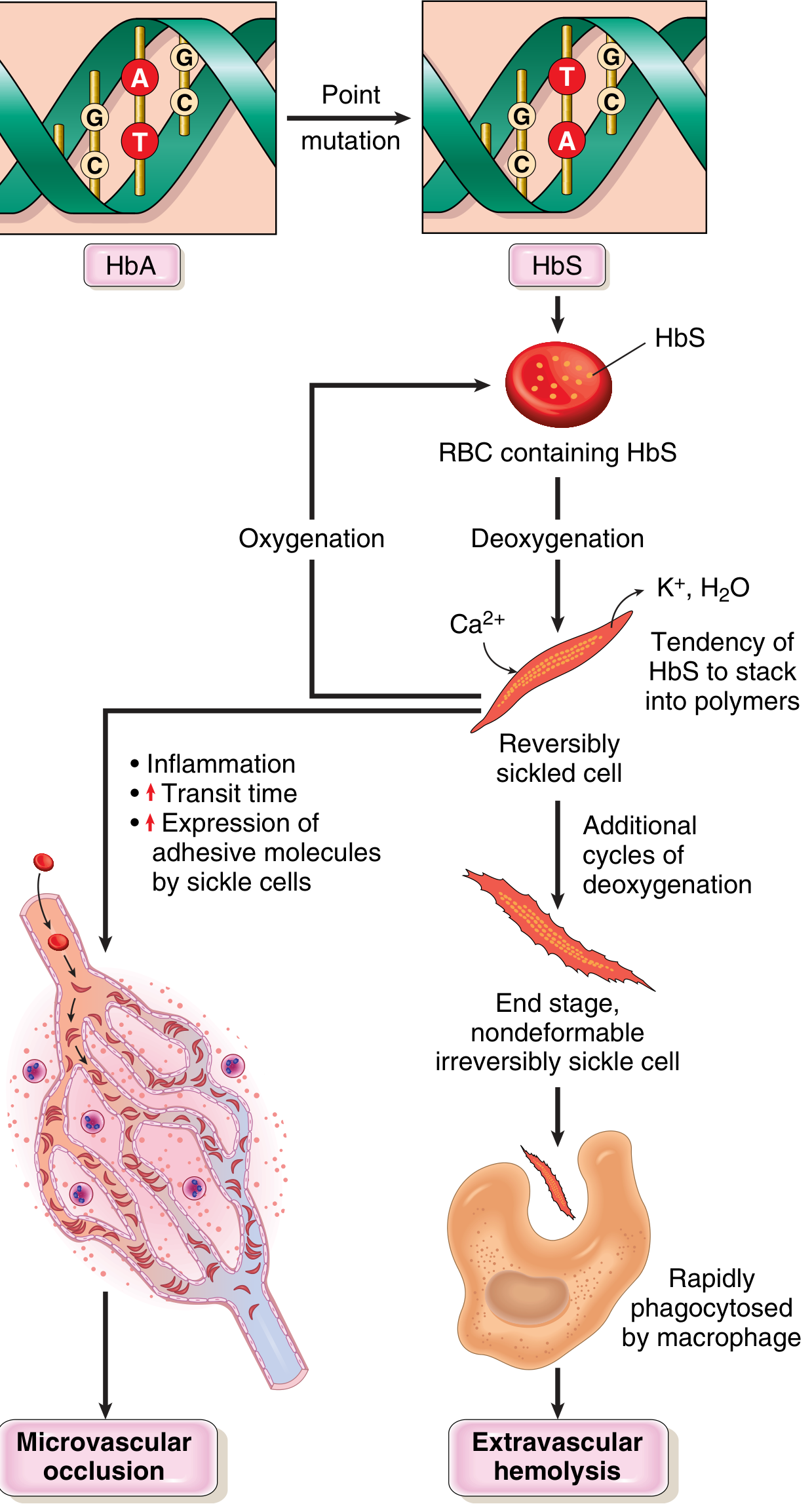
Step 1 - HbS polymerization on deoxygenation:
Deoxygenated HbS undergoes a conformational change that exposes the abnormal valine residue, allowing intermolecular contacts and polymer formation. These polymers distort the RBC into the classic elongated, crescentic sickle shape.
Step 2 - Sickling is initially reversible, then permanent:
- Reoxygenation initially reverses sickling
- Each sickling episode causes calcium influx → K⁺ and water loss → membrane skeleton damage
- Cumulative membrane damage creates irreversibly sickled cells prone to hemolysis
Three key determinants of sickling severity:
- Level of non-HbS hemoglobin - HbA and HbF both inhibit HbS polymerization. HbF is especially protective; this is why neonates are asymptomatic until ~5-6 months of age when HbF declines to adult levels.
- Intracellular HbS concentration - Dehydration raises intracellular Hb concentration and worsens sickling. Coexistent α-thalassemia (reduces Hb concentration) is actually protective.
- Microvascular transit time - Sluggish flow (spleen, bone marrow, inflamed tissues) allows sickling to occur. Sickled cells are abnormally "sticky," further prolonging transit time.
- Robbins & Kumar Basic Pathology, p. 386-387
Pathophysiology diagram:
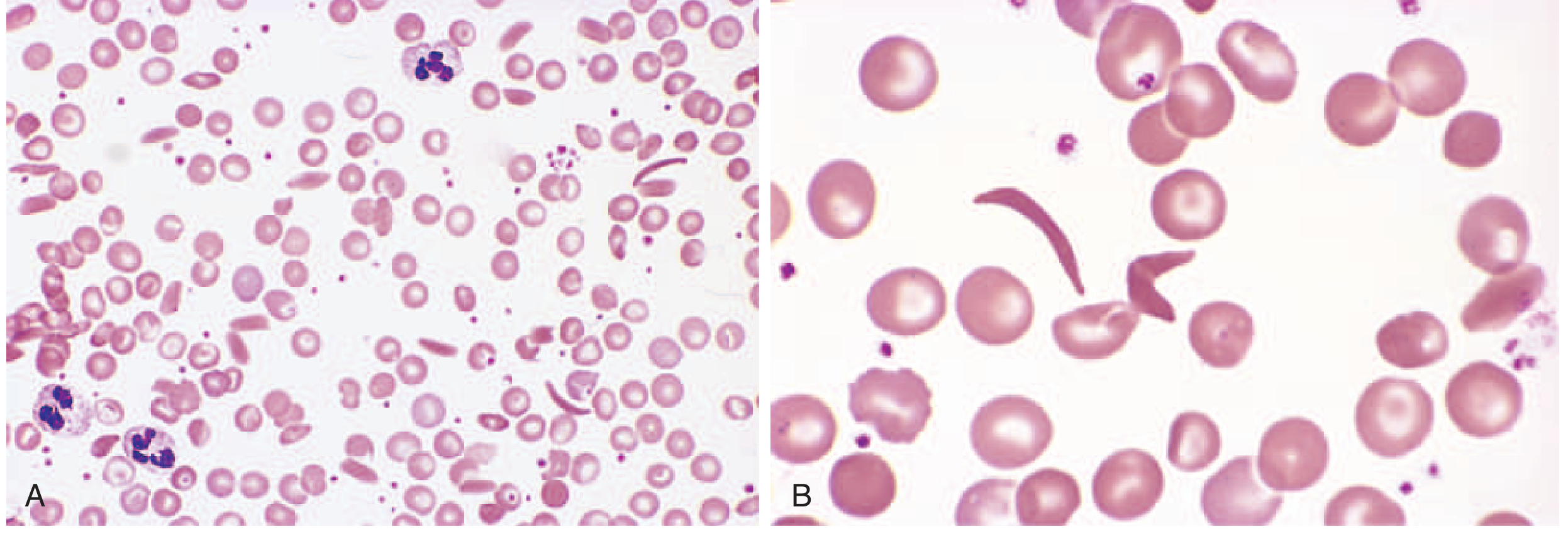
Peripheral Blood Smear
Elongated, spindled, boat-shaped sickled red cells are visible. Panel A shows a lower-power view of mixed cell morphology with some sickle forms; Panel B shows classic sickle cells at higher power.
Two Main Pathologic Consequences
1. Chronic Hemolytic Anemia
- Mean RBC lifespan: ~20 days (vs. ~120 days normal) - one-sixth of normal
- Hematocrit: typically 18-30% (normal 38-48%)
- Associated with hyperbilirubinemia, compensatory reticulocytosis, and compensatory erythroid hyperplasia in bone marrow
- Bone marrow expansion causes bone resorption → prominent cheekbones, "crew-cut" appearance on skull X-ray
- Extramedullary hematopoiesis may occur in liver and spleen
2. Vasoocclusive Crises
Triggered by superimposed factors: infection, inflammation, dehydration, acidosis. The number of irreversibly sickled cells does NOT correlate with vasoocclusion risk.
Clinical Features
Disease is asymptomatic until ~6 months of age (protected by fetal HbF).
Vasoocclusive Crises
| Crisis Type | Key Features |
|---|---|
| Hand-foot syndrome (dactylitis) | Most common presenting symptom in young children; infarction of small bones of hands/feet |
| Acute chest syndrome (ACS) | Sickling in hypoxemic pulmonary beds; may be triggered by pneumonia or fat emboli from infarcted bone; creates vicious cycle of hypoxemia → more sickling; leading cause of death |
| Stroke | Often occurs with ACS; leading cause of ischemia-related death |
| Proliferative retinopathy | Vasoocclusion in the eye → vision loss, blindness |
| Priapism | Frequent; can lead to penile fibrosis and impotence |
| Aplastic crisis | Sudden drop in RBC production; triggered by parvovirus B19 infecting erythroblasts; self-limited |
| Splenic sequestration | Acute pooling of blood in spleen; can cause life-threatening anemia in children |
| Bone infarction | Predisposes to osteomyelitis - most commonly Salmonella and encapsulated bacteria |
Organ Damage (Chronic)
-
Autosplenectomy by adulthood: spleen reduced to fibrotic nubbin; renders patients functionally asplenic and susceptible to encapsulated bacteria (pneumococci, H. influenzae, meningococci)
-
Hypoxia-induced fatty changes in heart, liver, renal tubules
-
Renal: glomerulosclerosis, papillary necrosis, concentrating defect (isosthenuria)
-
Chronic leg ulcers, avascular necrosis of femoral/humeral head
-
Pulmonary hypertension
-
Robbins & Kumar Basic Pathology, pp. 387-388
Diagnosis
- Newborn screening (mandatory in all 50 US states): Hemoglobin electrophoresis / HPLC on heel-stick blood
- Sickle preparation / Sickledex: Rapid tests positive for all sickle hemoglobinopathies (false negatives in neonates with high HbF)
- Prenatal diagnosis: Chorionic villus sampling or cell-free DNA testing; amniocentric fetal DNA analysis
- Transcranial Doppler (TCD): Screening begins at age 2 years, annually until age 16; velocity >200 cm/sec indicates high stroke risk requiring exchange transfusion
- Harriet Lane Handbook, 23rd ed.; Harrison's Principles 22e
Treatment
Disease-Modifying Drugs
Hydroxyurea (HU) - Standard of care
- Mechanism: Induces HbF synthesis (heterocellularly), reducing HbS polymerization
- Indicated for all patients with HbSS and HbSβ⁰ thalassemia, regardless of symptoms
- In adults: raises HbF from ~5% to ~10%, increases Hb ~1 g/dL, reduces pain crises and ACS by ~50%, and after 17.5 years follow-up, reduces mortality by 49%
- In infants started at <1 year (~27 mg/kg): HbF rises to ~33%, hemoglobin ~10 g/dL, acute events markedly reduced
- Current recommendation: Start at 9 months of age at ~20 mg/kg, titrated to maximum tolerated dose based on neutrophil and platelet counts
- Contraindicated in pregnancy (teratogenic)
Voxelotor
- Mechanism: Increases hemoglobin O₂ affinity, stabilizing the oxygenated conformation
- Dose 1500 mg/day: raises Hb ~1 g/dL in 59% of patients, reduces hemolysis biomarkers
- Additive to hydroxyurea; used when HU alone is insufficient
Crizanlizumab
- Mechanism: Anti-P-selectin monoclonal antibody; blocks sickle cell-endothelial adhesion
- Given IV monthly; in the pivotal trial, reduced acute painful episodes by ~45%
- A follow-up trial did not replicate these results; role remains uncertain
L-glutamine
- Reduces oxidative stress in sickle erythrocytes
- Phase 3 trial: 25% reduction in painful episodes, 33% reduction in hospitalization
Transfusion
- Simple transfusion for: symptomatic severe anemia, cardiopulmonary instability
- Exchange transfusion for: stroke, ACS with rapid progression/deterioration, multisystem organ failure, prior stroke, TCD >200 cm/sec
- Goal: lower HbS to <40%, target Hb ~10 g/dL; avoid hyperviscosity (keep Hct <30%)
- Prophylactic transfusions used in pregnancy to reduce vaso-occlusive crises
Curative Approaches
Hematopoietic stem cell transplantation (HSCT)
- HLA-identical related donor: event-free survival >95%
- Only ~15% of patients have a fully matched related donor
- Haploidentical transplant protocols improving outcomes
Gene Therapy (FDA-approved)
Two ex vivo approaches using autologous mobilized CD34+ stem cells, followed by myeloablative conditioning:
- Lentiviral vector introducing a modified HbA gene (BT87Q mutation) that inhibits HbS polymerization
- CRISPR/Cas9 disrupting BCL11A (the major HbF repressor), thereby reactivating HbF production Both achieve near-pancellular HbF or HbA^T87Q at 30-50%, total Hb >12 g/dL, and near-total prevention of acute vasoocclusive events. Long-term safety data are still accumulating.
- Harrison's Principles of Internal Medicine 22e (2025), p. 819
Preventive Care & Prophylaxis
| Measure | Details |
|---|---|
| Penicillin prophylaxis | 125 mg twice daily age <3 yr; 250 mg twice daily age 3-5 yr; continues until age 5 yr |
| Pneumococcal vaccines | PCV13 + PPSV23; critical given functional asplenia |
| Folic acid | 1 mg/day; prevents megaloblastic erythropoiesis |
| TCD screening | Annual from age 2-16 yr to detect stroke risk |
| Hydroxyurea | Start at 9 months in all HbSS/HbSβ⁰ infants |
Pregnancy Considerations
- Median life expectancy is now into the fifth decade; female fertility generally preserved
- Increased risk: preterm labor, PROM, infections, pain crises, thrombosis, pre-eclampsia, cesarean delivery
- 6-fold increased maternal mortality vs. controls
- Hydroxyurea is contraindicated in pregnancy (teratogenic)
- NSAIDs avoided after 30 weeks gestation
- Regional anesthesia preferred over general (reduces postpartum sickling complications)
- VTE risk 1.7-10x higher in pregnant SCD patients
- Rosen's Emergency Medicine, p. 3377
Summary of Key Facts
| Feature | Value |
|---|---|
| Mutation | β-globin codon 6: Glu → Val (GAG → GTG) |
| Inheritance | Autosomal recessive |
| RBC lifespan | ~20 days (1/6 normal) |
| Hematocrit | 18-30% |
| Age symptoms begin | ~6 months (as HbF declines) |
| Leading causes of death | Acute chest syndrome, stroke |
| Standard of care drug | Hydroxyurea |
| Infection prophylaxis | Penicillin until age 5 |
| Curative options | HSCT (>95% EFS with matched donor); gene therapy (lentiviral + CRISPR) |
This is a shared conversation. Sign in to Orris to start your own chat.