Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathophysiology
Reading File
Reading File
Searching Images
ARDS pathophysiology mechanism diffuse alveolar damage
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
I now have all the material needed for a comprehensive answer. Here is a full mechanistic explanation of ARDS:
Acute Respiratory Distress Syndrome — Mechanism
Definition and Diagnostic Criteria (Berlin 2012)
ARDS is defined by:
- Bilateral pulmonary infiltrates on chest X-ray or CT within 7 days of a known clinical insult
- PaO₂/FiO₂ (P/F) ratio <300 (mild <300, moderate <200, severe <100)
- Non-cardiogenic pulmonary edema (not explained by heart failure or fluid overload)
- Positive end-expiratory pressure ≥5 cm H₂O on mechanical or non-invasive ventilation
— Sabiston Textbook of Surgery, p. 940
Precipitating Causes
Injury reaches the lung by two routes:
| Direct (Pulmonary) | Indirect (Extrapulmonary) |
|---|---|
| Pneumonia | Sepsis (most common overall) |
| Aspiration | Major trauma / burns |
| Pulmonary contusion | Pancreatitis |
| Toxic inhalation | Massive transfusion (TRALI) |
| Near-drowning | Hemorrhagic shock |
Direct ARDS produces more radiographic heterogeneity, lower compliance, and higher biomarkers of epithelial injury. Indirect ARDS tends toward radiographic homogeneity, higher compliance, and higher biomarkers of endothelial injury.
— Fishman's Pulmonary Diseases and Disorders, p. 2494
Core Pathophysiologic Mechanism
ARDS is fundamentally an uncontrolled, self-amplifying inflammatory response to injury of the alveolar–capillary unit. The full sequence unfolds in two overlapping phases.
Phase 1 — Exudative Phase (Hours → ~7 days)
Step 1: Initiating Injury — Pneumocyte and Endothelial Damage
The cascade begins with injury to either type I alveolar pneumocytes or pulmonary capillary endothelium (or both simultaneously in sepsis/shock).
- Resident alveolar macrophages detect pneumocyte injury via pattern-recognition receptors and release TNF-α, IL-1β, IL-6, IL-8, and other pro-inflammatory mediators.
- In systemic insults (sepsis, trauma), circulating mediators — LPS, damage-associated molecular patterns (DAMPs), complement fragments — activate pulmonary endothelium directly, independent of pneumocyte injury.
— Robbins & Cotran Pathologic Basis of Disease, p. 632
Step 2: Endothelial Activation and Neutrophil Recruitment
Cytokines (TNF-α, IL-1β) induce endothelial cells to upregulate:
- Adhesion molecules (ICAM-1, E-selectin, P-selectin) → neutrophil margination and rolling
- Chemokines (IL-8/CXCL8) → neutrophil transmigration
- Procoagulant proteins → local microvascular thrombosis
Neutrophils adhere to activated endothelium, extravasate into the interstitium and alveolar spaces, and then degranulate, releasing:
- Proteases (elastase, matrix metalloproteinases) — destroy extracellular matrix, tight junctions, and the alveolar basement membrane
- Reactive oxygen species (ROS) — oxidative damage to lipid membranes and surfactant
- Neutrophil extracellular traps (NETs) — directly damage alveolar epithelium
- Additional cytokines — positive-feedback amplification of the inflammatory cascade
This neutrophil-driven injury is the central effector mechanism of alveolar damage in ARDS.
— Robbins & Cotran Pathologic Basis of Disease, p. 632
Step 3: Breakdown of the Alveolar–Capillary Barrier
The combined assault on endothelial tight junctions and the alveolar epithelium results in a massive increase in capillary permeability. This allows protein-rich plasma to pour across the broken barrier into:
- The interstitium → interstitial edema, widened intercellular junctions (visible on electron microscopy as endothelial swelling)
- The alveolar space → alveolar flooding with proteinaceous edema fluid, fibrin, and cellular debris
Early on (hours), electron microscopy shows endothelial swelling, widening of intercellular junctions, increased pinocytotic vesicles, and disruption and denudation of the basement membrane. Light microscopy then shows interstitial and alveolar edema, capillary congestion, and intra-alveolar hemorrhage.
— Fishman's Pulmonary Diseases and Disorders, p. 2494
Step 4: Surfactant Inactivation and Alveolar Collapse
- Alveolar flooding washes out and inactivates surfactant (phospholipase A2 released during pancreatitis/systemic inflammation also directly degrades surfactant phospholipids)
- Loss of surfactant eliminates surface tension reduction → alveolar instability and collapse (atelectasis)
- Type II pneumocyte necrosis further depletes surfactant production capacity
— Fishman's Pulmonary Diseases and Disorders, p. 2495; Murray & Nadel's, p. 2961
Step 5: Hyaline Membrane Formation
Over the next few days, inspissated protein-rich edema fluid, condensed fibrin, and cellular debris from necrotic pneumocytes organize on the denuded alveolar walls to form hyaline membranes — the pathognomonic histologic feature of diffuse alveolar damage (DAD).
— Goldman-Cecil Medicine, p. 927; Robbins & Cotran, p. 632
The Diagram — Normal vs. Injured Alveolus (Robbins & Cotran)
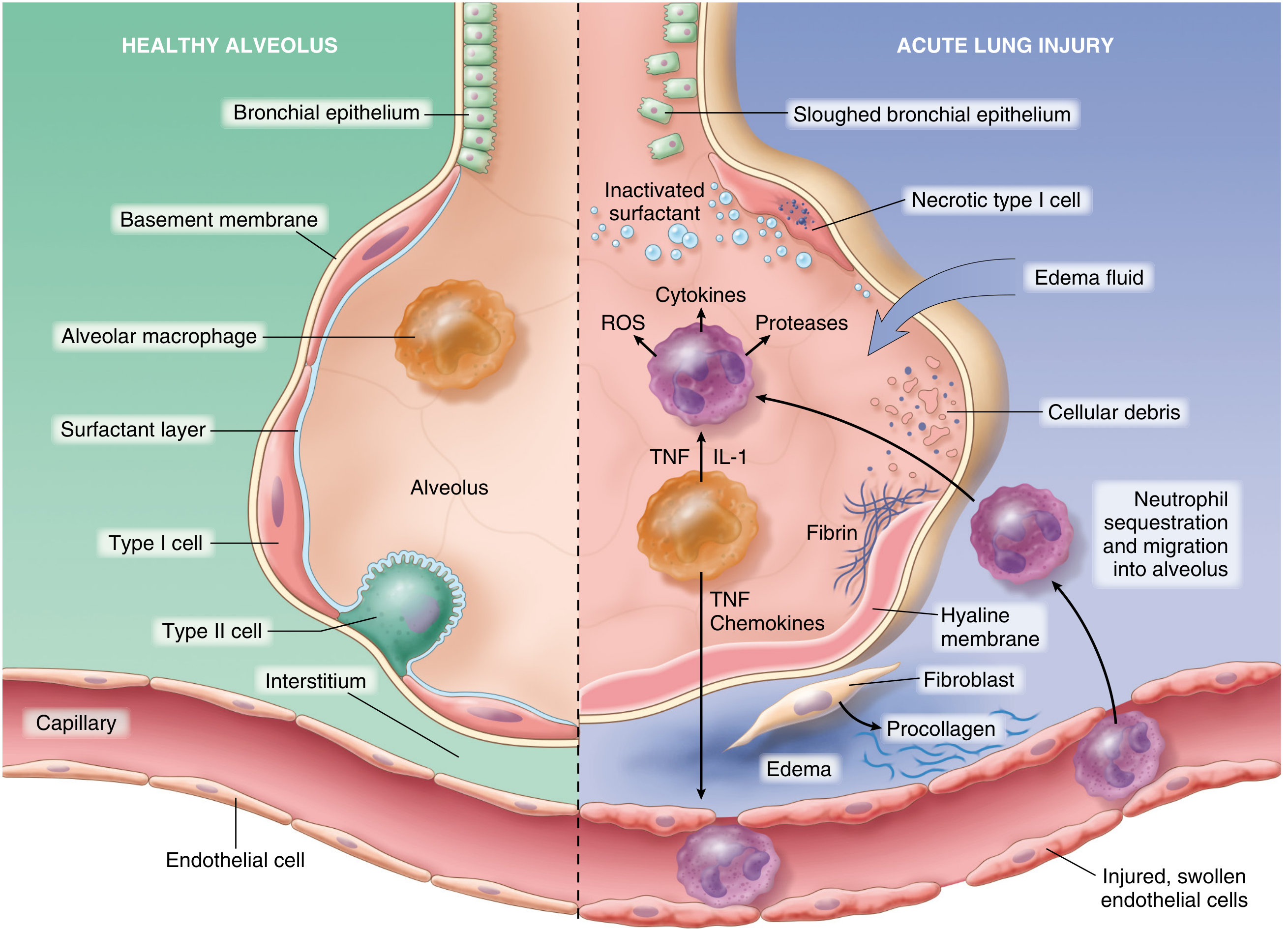
Fig. 15.3 — The normal alveolus (left) compared with the injured alveolus (right) in the early phase of ALI/ARDS. (Robbins, Cotran & Kumar Pathologic Basis of Disease)
Histopathology of DAD and CT Correlation
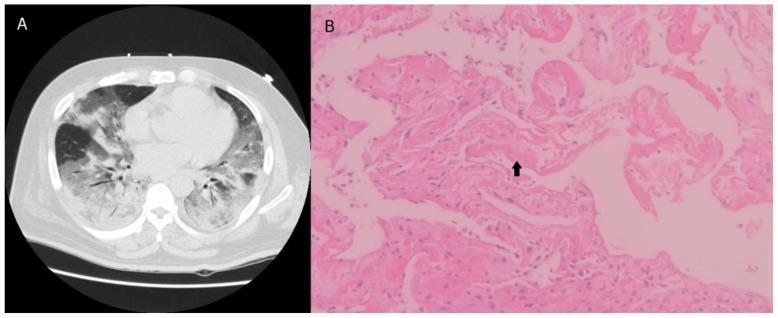
Panel A: Chest CT showing bilateral ground-glass opacities and consolidation. Panel B: H&E histology showing hyaline membranes (arrow) and alveolar wall thickening — hallmarks of DAD in ARDS.
Gas Exchange Failure — The Physiology
Three mechanisms converge to produce the characteristic refractory hypoxemia:
1. Intrapulmonary Shunt (Dominant Mechanism)
- Flooded and collapsed alveoli receive no ventilation but continue to be perfused → V/Q ratio = 0 → blood passes through the lung without oxygenation (true right-to-left shunt)
- In early MIGET (multiple inert gas elimination technique) studies of ARDS, up to 48% of cardiac output flowed through shunt or very low V/Q areas
- Shunt is refractory to supplemental O₂ — increasing FiO₂ has little effect because the shunted blood never contacts the alveolar gas
- PEEP reduces shunt by recruiting collapsed/flooded alveoli, redistributing blood to ventilated zones
2. V/Q Mismatch
- Heterogeneous distribution of edema and atelectasis creates areas of very low (but non-zero) V/Q that add shunt-like contributions to hypoxemia
3. Reduced Lung Compliance
- Fluid-filled, atelectatic lung has dramatically decreased compliance (ΔV/ΔP)
- Increased work of breathing, tendency to small tidal volumes
- Gravity-dependent consolidation ("sponge lung" model): nondependent zones retain near-normal compliance while dependent zones are flooded/atelectatic
— Fishman's Pulmonary Diseases and Disorders, p. 2495; Murray & Nadel's, p. 4419–4421
Phase 2 — Fibroproliferative (Organizing/Late) Phase (~7–14+ days)
If the initial inflammatory insult persists or resolution is impaired:
- Macrophages clear intra-alveolar debris and release fibrogenic cytokines: TGF-β and PDGF
- TGF-β and PDGF stimulate fibroblast and myofibroblast proliferation → collagen deposition → alveolar wall fibrosis
- Type II pneumocytes proliferate to replace necrotic type I cells (type II hyperplasia is a histologic hallmark of this phase)
- Alveolar septal thickening, disordered angiogenesis, and pulmonary hypertension develop
- Dead-space fraction increases (pulmonary vascular obliteration + microthrombi)
- High minute ventilation requirement reflects increased dead space
- Lung compliance falls further
— Fishman's Pulmonary Diseases and Disorders, p. 2495; Robbins & Cotran, p. 632
ARDS Endotypes (Modern Concept)
Latent class analysis of large ARDS Network trial datasets has identified two biologically distinct endotypes:
| Feature | Hyperinflammatory (Endotype 2) | Hypoinflammatory (Endotype 1) |
|---|---|---|
| Plasma IL-6, IL-8, TNF-α | Very high | Low |
| Shock | More common | Less common |
| Mortality | Higher | Lower |
| Response to PEEP | May differ | May differ |
This endotyping suggests ARDS is not a single disease but a heterogeneous syndrome sharing a final common pathway of diffuse alveolar damage.
— Fishman's Pulmonary Diseases and Disorders, p. 2494
Coagulation Dysregulation
A frequently overlooked component: injury activates the coagulation cascade within alveoli and microvasculature:
- Exposed tissue factor on damaged endothelium → thrombin generation → intravascular fibrin microthrombi
- Simultaneous depression of fibrinolysis (raised PAI-1)
- Intra-alveolar fibrin contributes to hyaline membrane formation and impairs resolution
- Small pulmonary artery in situ thrombi → pulmonary hypertension and increased dead space
— Goldman-Cecil Medicine, p. 927
Ventilatior-Induced Lung Injury (VILI) — A Secondary Mechanism
Mechanical ventilation itself can perpetuate ARDS injury via:
- Volutrauma / Barotrauma — overdistension of non-dependent alveoli at high tidal volumes or pressures
- Atelectrauma — repeated collapse-reopening of unstable alveoli
- Biotrauma — mechanical strain releases cytokines/mediators from alveolar cells into the bloodstream → distal organ dysfunction (kidney, gut, brain)
Lung-protective ventilation (6 mL/kg tidal volume, plateau pressure ≤30 cmH₂O) attenuates both volutrauma and biotrauma and reduces mortality.
— Goldman-Cecil Medicine, p. 1066–1067
Summary Cascade
Triggering insult (sepsis/pneumonia/trauma/etc.)
↓
Alveolar macrophage activation → TNF-α, IL-1β, IL-8
↓
Endothelial upregulation of adhesion molecules
↓
Neutrophil adhesion → transmigration into alveoli
↓
Release of proteases + ROS + NETs
↓
Destruction of tight junctions + basement membrane
↓
Protein-rich flood into alveolus → surfactant washout/inactivation
↓
Alveolar collapse + hyaline membrane formation (DAD)
↓
Massive right-to-left shunt → refractory hypoxemia
↓
If sustained → fibroproliferative repair → fibrosis + pulmonary HTN
Key sources:
- Robbins, Cotran & Kumar Pathologic Basis of Disease (Fig. 15.3 and surrounding text)
- Fishman's Pulmonary Diseases and Disorders (Chapter 141)
- Murray & Nadel's Textbook of Respiratory Medicine (Chapters 44, 126)
- Goldman-Cecil Medicine (Chapters 79, 91)
- Sabiston Textbook of Surgery (Chapter 11)
This is a shared conversation. Sign in to Orris to start your own chat.